
Insulin Resistance and Hypertension: Mechanisms Involved and Modifying Factors for Effective Glucose Control

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Hypertension: The Silent Killer
2. Insulin Resistance
3. Diet-Induced Insulin Resistance
3.1. Fructose
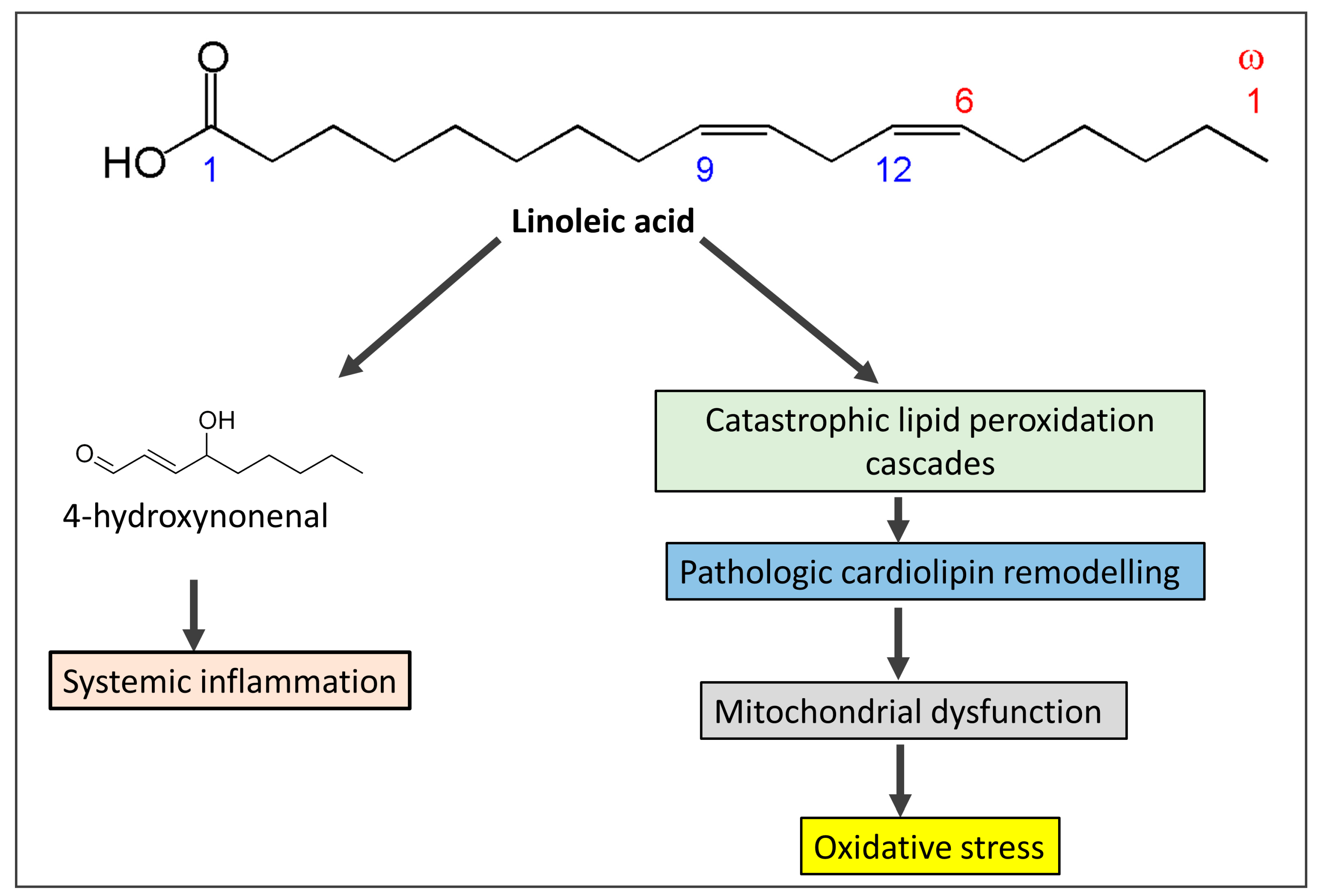
3.2. Omega-6 Fatty Acids
3.3. Grains
4. The Detailed Mechanism of Insulin Resistance Linked to Hypertension
4.1. Advanced Glycation End Products (AGEs) and Hypertension
4.2. Oxidative Stress and Hypertension
4.3. Systemic Inflammation and Hypertension
4.4. Dyslipidemia and Hypertension
4.5. Endothelial Dysfunction and Hypertension
4.6. Hypervolemia and Hypertension
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC-1 | Acetyl-CoA carboxylase |
| ACEs | Angiotensin Converting Enzymes |
| AGEs | Advanced Glycation Endproducts |
| AMPD | AMP deaminase |
| AP1 | Activator protein 1 |
| ApoB | Apolipoprotein B |
| AT1R | Angiotensin type 1 receptor |
| BMI | Body mass index |
| CAD | Coronary artery disease |
| CPT-1 | Carntine Palmyle transferase |
| CRP | C reactive protein |
| DHEA | Dehydroepiandrosterone |
| DNA | Deoxyribonucleic acid |
| DNL | De novo lipogensesis |
| ECM | Extracellular matrices |
| F1P | Fructose-1-phosphate |
| FAS | Fatty acid synthase |
| FFA | Free fatty acids |
| Glut 4 | Glucose transporter type 4 |
| HDL | High-density lipoprotein |
| Hepatic DNL | Hepatic de novo lipogenesis |
| HFCS | High fructose corn syrup |
| HICs | High-income countries |
| HMGCOAR | Hydroxymethylglutaryl-coenzyme A reductase |
| HMGCOAS | Hydroxymethylglutaryl-coenzyme A synthase |
| IL-17 | Interleukin 17 |
| IL-6 | Interleukin 6 |
| IRS1 | Insulin resistance substrate 1 |
| IR | Insulin resistance |
| IRS-1 | Insulin receptor substrate-1 |
| KLF2/4 | Kruppel-like factor |
| LCFH | Low-carb, high-fat |
| MAPKs | Mitogen-activated protein kinases |
| MetS | Metabolic syndrome |
| MMP | Matrix metalloproteinase |
| mRNA | MicroRNA |
| MT1 | Membrane type-1 |
| Na | sodium |
| NAFLD | Non-alcoholic fatty liver disease |
| NFβ | Nuclear factor-κB |
| NHE3 | Na+-H+ exchanger type 3 |
| NO | Nitric oxide |
| p-ACC-1 | Phospho-Acetyl-CoA Carboxylase |
| p-AMPK | Phosphorylated adenosine monokinase |
| PCOS | Polycystic ovarioan syndrome |
| PDGF | Platelet derived growth factor |
| PECAM 1 | Platelet endothelial cell adhesion molecule-1 |
| PKC | Protein kinase C |
| PPAR-α | Peroxisome proliferator-activated receptor alpha |
| p-PPAR-α, | Phosphorylated Peroxisome proliferator-activated receptor alpha |
| RAAS | Renin angiotensin aldosterone system |
| ROS | Reactive oxygen species |
| SHBG | Sex hormone binding globulin |
| SREBP-1 | Sterol Regulatory Element-binding Protein-1 |
| SREBP-2 | Sterol regulatory element-binding protein 2 |
| T2DM | Type 2 diabetes mellitus |
| TNF-α | Tumour necrosis factor alpha |
| tPA | Tissue plasminogen activator |
| TSH | Thyroid stimulating hormone |
| UCP-2 | Uncoupling protein-2 |
| u-PA | Urokinase plasminogen activator |
| VLDL | Very low-density lipoprotein |
| VSMC | Vascular smooth muscle cell |
| WGA | Wheat germ agglutinin |
References
- GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the global burden of disease study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar]
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the global burden of disease study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global disparities of hypertension prevalence and control: A systematic analysis of population-based studies from 90 countries. Circulation 2016, 134, 441–450. [Google Scholar]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [PubMed]
- Pokharel, Y.; Karmacharya, B.M.; Neupane, D. Hypertension—A silent killer without global bounds: What next? J. Am. Coll. Cardiol. 2022, 80, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Suvila, K.; Langén, V.; Cheng, S.; Niiranen, T.J. Age of hypertension onset: Overview of research and how to apply in practice. Curr. Hypertens. Rep. 2020, 22, 020–01071. [Google Scholar]
- Sica, D.A. Endocrine causes of secondary hypertension. J. Clin. Hypertens. 2008, 10, 534–540. [Google Scholar]
- Gupta, R.; Guptha, S. Strategies for initial management of hypertension. Indian J. Med. Res. 2010, 132, 531–542. [Google Scholar]
- Williams, B. Treating hypertension: It is not how you start but where you end that matters. J. Hypertens. 2003, 21, 455–457. [Google Scholar]
- Ivy, J.L. Muscle insulin resistance amended with exercise training: Role of glut4 expression. Med. Sci. Sports Exerc. 2004, 36, 1207–1211. [Google Scholar]
- Bremer, A.A.; Mietus-Snyder, M.; Lustig, R.H. Toward a unifying hypothesis of metabolic syndrome. Pediatrics 2012, 129, 557–570. [Google Scholar] [PubMed]
- Roden, M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol. Sci. 2004, 19, 92–96. [Google Scholar] [PubMed]
- Honzawa, N.; Fujimoto, K.; Kitamura, T. Cell autonomous dysfunction and insulin resistance in pancreatic α cells. Int. J. Mol. Sci. 2019, 20, 3699. [Google Scholar]
- Li, Q.; Zhao, M.; Wang, Y.; Zhong, F.; Liu, J.; Gao, L.; Zhao, J. Associations between serum free fatty acid levels and incident diabetes in a 3-year cohort study. Diabetes Metab. Syndr. Obes. 2021, 14, 2743–2751. [Google Scholar] [PubMed]
- Sabbatini, A.R.; Fontana, V.; Laurent, S.; Moreno, H. An update on the role of adipokines in arterial stiffness and hypertension. J. Hypertens. 2015, 33, 435–444. [Google Scholar]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar]
- Sakr, H.F.; Sirasanagandla, S.R.; Das, S.; Bima, A.I.; Elsamanoudy, A.Z. Low-carbohydrate ketogenic diet for improvement of glycemic control: Mechanism of action of ketosis and beneficial effects. Curr. Diabetes Rev. 2022, 11. [Google Scholar] [CrossRef]
- Rabe, K.; Lehrke, M.; Parhofer, K.G.; Broedl, U.C. Adipokines and insulin resistance. Mol. Med. 2008, 14, 741–751. [Google Scholar]
- Pankow, J.S.; Jacobs, D.R., Jr.; Steinberger, J.; Moran, A.; Sinaiko, A.R. Insulin resistance and cardiovascular disease risk factors in children of parents with the insulin resistance (metabolic) syndrome. Diabetes Care 2004, 27, 775–780. [Google Scholar]
- Vaag, A.; Henriksen, J.E.; Madsbad, S.; Holm, N.; Beck-Nielsen, H. Insulin secretion, insulin action, and hepatic glucose production in identical twins discordant for non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1995, 95, 690–698. [Google Scholar]
- Gerich, J.E. The genetic basis of type 2 diabetes mellitus: Impaired insulin secretion versus impaired insulin sensitivity. Endocr. Rev. 1998, 19, 491–503. [Google Scholar] [PubMed]
- Chiu, K.C.; Cohan, P.; Lee, N.P.; Chuang, L.M. Insulin sensitivity differs among ethnic groups with a compensatory response in beta-cell function. Diabetes Care 2000, 23, 1353–1358. [Google Scholar] [PubMed]
- Nuwaylati, D.; Eldakhakhny, B.; Bima, A.; Sakr, H.; Elsamanoudy, A. Low-carbohydrate high-fat diet: A swoc analysis. Metabolites 2022, 12, 1126. [Google Scholar]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar]
- Maggio, M.; Lauretani, F.; Ceda, G.P.; Bandinelli, S.; Ling, S.M.; Metter, E.J.; Artoni, A.; Carassale, L.; Cazzato, A.; Ceresini, G.; et al. Relationship between low levels of anabolic hormones and 6-year mortality in older men: The aging in the chianti area (inchianti) study. Arch. Intern. Med. 2007, 167, 2249–2254. [Google Scholar] [PubMed]
- Muller, M.; Grobbee, D.E.; den Tonkelaar, I.; Lamberts, S.W.; van der Schouw, Y.T. Endogenous sex hormones and metabolic syndrome in aging men. J. Clin. Endocrinol. Metab. 2005, 90, 2618–2623. [Google Scholar] [CrossRef]
- Kapoor, D.; Goodwin, E.; Channer, K.S.; Jones, T.H. Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur. J. Endocrinol. 2006, 154, 899–906. [Google Scholar]
- Sakr, H.F.; Hussein, A.M.; Eid, E.A.; AlKhateeb, M. Possible mechanisms underlying fatty liver in a rat model of male hypogonadism: A protective role for testosterone. Steroids 2018, 135, 21–30. [Google Scholar] [PubMed]
- Asfari, M.M.; Sarmini, M.T.; Baidoun, F.; Al-Khadra, Y.; Ezzaizi, Y.; Dasarathy, S.; McCullough, A. Association of non-alcoholic fatty liver disease and polycystic ovarian syndrome. BMJ Open Gastroenterol. 2020, 7, e000352. [Google Scholar]
- Song, M.J.; Choi, J.Y. Androgen dysfunction in non-alcoholic fatty liver disease: Role of sex hormone binding globulin. Front. Endocrinol. 2022, 13, 1053709. [Google Scholar]
- Verma, N.; Jain, V.; Birla, S.; Jain, R.; Sharma, A. Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J. Pediatr. Endocrinol. Metab. 2012, 25, 1185–1190. [Google Scholar]
- Salpeter, S.R.; Walsh, J.M.; Ormiston, T.M.; Greyber, E.; Buckley, N.S.; Salpeter, E.E. Meta-analysis: Effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes. Metab. 2006, 8, 538–554. [Google Scholar] [PubMed]
- Deibert, D.C.; DeFronzo, R.A. Epinephrine-induced insulin resistance in man. J. Clin. Investig. 1980, 65, 717–721. [Google Scholar] [PubMed]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [PubMed]
- Bastemir, M.; Akin, F.; Alkis, E.; Kaptanoglu, B. Obesity is associated with increased serum tsh level, independent of thyroid function. Swiss Med. Wkly. 2007, 137, 431–434. [Google Scholar]
- Reinehr, T.; Andler, W. Thyroid hormones before and after weight loss in obesity. Arch. Dis. Child. 2002, 87, 320–323. [Google Scholar]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar]
- Weickert, M.O. Nutritional modulation of insulin resistance. Scientifica 2012, 2012, 424780. [Google Scholar]
- Shan, Z.; Rehm, C.D.; Rogers, G.; Ruan, M.; Wang, D.D.; Hu, F.B.; Mozaffarian, D.; Zhang, F.F.; Bhupathiraju, S.N. Trends in dietary carbohydrate, protein, and fat intake and diet quality among us adults, 1999–2016. JAMA 2019, 322, 1178–1187. [Google Scholar]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [PubMed]
- Serna-Saldivar, S.O. Maize: Foods from Maize. Ref. Modul. Food Sci. 2016. [Google Scholar] [CrossRef]
- Pereira, R.M.; Botezelli, J.D.; da Cruz Rodrigues, K.C.; Mekary, R.A.; Cintra, D.E.; Pauli, J.R.; da Silva, A.S.R.; Ropelle, E.R.; de Moura, L.P. Fructose consumption in the development of obesity and the effects of different protocols of physical exercise on the hepatic metabolism. Nutrients 2017, 9, 405. [Google Scholar] [PubMed]
- Marriott, B.P.; Olsho, L.; Hadden, L.; Connor, P. Intake of added sugars and selected nutrients in the united states, national health and nutrition examination survey (nhanes) 2003–2006. Crit. Rev. Food Sci. Nutr. 2010, 50, 228–258. [Google Scholar] [PubMed]
- Tappy, L.; Lê, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of nafld and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Fructose selectively modulates c-jun n-terminal kinase activity and insulin signaling in rat primary hepatocytes. J. Nutr. 2005, 135, 1642–1646. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Fructose-mediated stress signaling in the liver: Implications for hepatic insulin resistance. J. Nutr. Biochem. 2007, 18, 1–9. [Google Scholar]
- Hallfrisch, J. Metabolic effects of dietary fructose. Faseb J. 1990, 4, 2652–2660. [Google Scholar] [CrossRef]
- Smith, C.M.; Rovamo, L.M.; Raivio, K.O. Fructose-induced adenine nucleotide catabolism in isolated rat hepatocytes. Can. J. Biochem. 1977, 55, 1237–1240. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y.; Cheng, S.; Sun, J.L.; Yao, H.; Ma, L. Effect of uric acid on mitochondrial function and oxidative stress in hepatocytes. Genet. Mol. Res. 2016, 15, 15028644. [Google Scholar] [CrossRef] [PubMed]
- Cicerchi, C.; Li, N.; Kratzer, J.; Garcia, G.; Roncal-Jimenez, C.A.; Tanabe, K.; Hunter, B.; Rivard, C.J.; Sautin, Y.Y.; Gaucher, E.A.; et al. Uric acid-dependent inhibition of amp kinase induces hepatic glucose production in diabetes and starvation: Evolutionary implications of the uricase loss in hominids. Faseb J. 2014, 28, 3339–3350. [Google Scholar]
- Softic, S.; Stanhope, K.L.; Boucher, J.; Divanovic, S.; Lanaspa, M.A.; Johnson, R.J.; Kahn, C.R. Fructose and hepatic insulin resistance. Crit. Rev. Clin. Lab. Sci. 2020, 57, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Whaley-Connell, A.T.; Sowers, J.R. Fructose and uric acid: Is there a role in endothelial function? Curr. Hypertens. Rep. 2014, 16, 014–0434. [Google Scholar]
- DiNicolantonio, J.J.; O’Keefe, J.H. Omega-6 vegetable oils as a driver of coronary heart disease: The oxidized linoleic acid hypothesis. Open Heart 2018, 5, e000898. [Google Scholar]
- Fritsche, K.L. Linoleic acid, vegetable oils & inflammation. Mo. Med. 2014, 111, 41–43. [Google Scholar]
- Aglago, E.K.; Biessy, C.; Torres-Mejía, G.; Angeles-Llerenas, A.; Gunter, M.J.; Romieu, I.; Chajès, V. Association between serum phospholipid fatty acid levels and adiposity in Mexican women. J. Lipid Res. 2017, 58, 1462–1470. [Google Scholar] [CrossRef]
- Mercola, J.; D’Adamo, C.R. Linoleic Acid: A Narrative Review of the Effects of Increased Intake in the Standard American Diet and Associations with Chronic Disease. Nutrients 2023, 15, 3129. [Google Scholar]
- Simopoulos, A.P. An increase in the omega-6/omega-3 fatty acid ratio increases the risk for obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- Gibson, R.A.; Muhlhausler, B.; Makrides, M. Conversion of linoleic acid and alpha-linolenic acid to long-chain polyunsaturated fatty acids (lcpufas), with a focus on pregnancy, lactation and the first 2 years of life. Matern. Child. Nutr. 2011, 2, 17–26. [Google Scholar] [CrossRef]
- Schwertner, H.A.; Mosser, E.L. Comparison of lipid fatty acids on a concentration basis vs. weight percentage basis in patients with and without coronary artery disease or diabetes. Clin. Chem. 1993, 39, 659–663. [Google Scholar] [CrossRef]
- Hodgson, J.M.; Wahlqvist, M.L.; Boxall, J.A.; Balazs, N.D. Can linoleic acid contribute to coronary artery disease? Am. J. Clin. Nutr. 1993, 58, 228–234. [Google Scholar] [CrossRef] [PubMed]
- El-Hafidi, M.; Correa, F.; Zazueta, C. Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165744. [Google Scholar] [CrossRef] [PubMed]
- Abuajah, C.I.; Ogbonna, A.C.; Osuji, C.M. Functional components and medicinal properties of food: A review. J. Food Sci. Technol. 2015, 52, 2522–2529. [Google Scholar] [CrossRef]
- Thomas, M.C.; Tikellis, C.; Burns, W.M.; Bialkowski, K.; Cao, Z.; Coughlan, M.T.; Jandeleit-Dahm, K.; Cooper, M.E.; Forbes, J.M. Interactions between renin angiotensin system and advanced glycation in the kidney. J. Am. Soc. Nephrol. 2005, 16, 2976–2984. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef]
- Sehgel, N.L.; Sun, Z.; Hong, Z.; Hunter, W.C.; Hill, M.A.; Vatner, D.E.; Vatner, S.F.; Meininger, G.A. Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension 2015, 65, 370–377. [Google Scholar] [CrossRef]
- Wirth, A.; Wang, S.; Takefuji, M.; Tang, C.; Althoff, T.F.; Schweda, F.; Wettschureck, N.; Offermanns, S. Age-dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by g-protein signalling. Cardiovasc. Res. 2016, 109, 131–140. [Google Scholar] [CrossRef]
- Drewnowski, A.; Krahn, D.D.; Demitrack, M.A.; Nairn, K.; Gosnell, B.A. Naloxone, an opiate blocker, reduces the consumption of sweet high-fat foods in obese and lean female binge eaters. Am. J. Clin. Nutr. 1995, 61, 1206–1212. [Google Scholar] [CrossRef]
- Winkler, S.; Kaplan, D.L. Biosynthesized Materials: Properties and Processing. In Encyclopedia of Materials: Science and Technology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 609–615. [Google Scholar] [CrossRef]
- Farrell, J.J. Digestion and Absorption of Nutrients and Vitamins. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Roriz-Filho, J.S.; Sá-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2009, 1792, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Urade, R.; Sato, N.; Sugiyama, M. Gliadins from wheat grain: An overview, from primary structure to nanostructures of aggregates. Biophys. Rev. 2018, 10, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, S.W.; Sun, Q.; Xia, G.M. High frequency of hmw-gs sequence variation through somatic hybridization between agropyron elongatum and common wheat. Planta 2010, 231, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Xia, X.C.; He, Z.H.; Gale, K.R.; Lei, Z.S.; Appels, R.; Ma, W. Characterization of three low-molecular-weight glu-d3 subunit genes in common wheat. Theor. Appl. Genet. 2006, 113, 1247–1259. [Google Scholar] [CrossRef]
- Monsigny, M.; Roche, A.C.; Sene, C.; Maget-Dana, R.; Delmotte, F. Sugar-lectin interactions: How does wheat-germ agglutinin bind sialoglycoconjugates? Eur. J. Biochem. 1980, 104, 147–153. [Google Scholar] [CrossRef]
- Peumans, W.J.; Stinissen, H.M.; Carlier, A.R. Isolation and partial characterization of wheat-germ-agglutinin-like lectins from rye (Secale cereale) and barley (Hordeum vulgare) embryos. Biochem. J. 1982, 203, 239–243. [Google Scholar] [CrossRef]
- Hamid, R.; Masood, A. Dietary lectins as disease causing toxicants. Pak. J. Nutr. 2009, 8, 293–303. [Google Scholar] [CrossRef]
- Lorenzsonn, V.; Olsen, W.A. In Vivo responses of rat intestinal epithelium to intraluminal dietary lectins. Gastroenterology 1982, 82, 838–848. [Google Scholar] [CrossRef]
- Longin, C.F.H.; Afzal, M.; Pfannstiel, J.; Bertsche, U.; Melzer, T.; Ruf, A.; Heger, C.; Pfaff, T.; Schollenberger, M.; Rodehutscord, M. Mineral and phytic acid content as well as phytase activity in flours and breads made from different wheat species. Int. J. Mol. Sci. 2023, 24, 2770. [Google Scholar] [CrossRef]
- Al Hasan, S.M.; Hassan, M.; Saha, S.; Islam, M.; Billah, M.; Islam, S. Dietary phytate intake inhibits the bioavailability of iron and calcium in the diets of pregnant women in rural bangladesh: A cross-sectional study. BMC Nutr. 2016, 2, 24. [Google Scholar] [CrossRef]
- James, J.M.; Sixbey, J.P.; Helm, R.M.; Bannon, G.A.; Burks, A.W. Wheat alpha-amylase inhibitor: A second route of allergic sensitization. J. Allergy Clin. Immunol. 1997, 99, 239–244. [Google Scholar] [CrossRef]
- Pastorello, E.A.; Farioli, L.; Conti, A.; Pravettoni, V.; Bonomi, S.; Iametti, S.; Fortunato, D.; Scibilia, J.; Bindslev-Jensen, C.; Ballmer-Weber, B.; et al. Wheat ige-mediated food allergy in european patients: Alpha-amylase inhibitors, lipid transfer proteins and low-molecular-weight glutenins. Allergenic molecules recognized by double-blind, placebo-controlled food challenge. Int. Arch. Allergy Immunol. 2007, 144, 10–22. [Google Scholar] [PubMed]
- Sinha, S.; Haque, M. Insulin resistance is cheerfully hitched with hypertension. Life 2022, 12, 564. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Baynes, J.W. Glycation. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Katta, R. Sugar sag: Glycation and the role of diet in aging skin. Ski. Ther. Lett. 2015, 20, 1–5. [Google Scholar]
- Kim, C.S.; Park, S.; Kim, J. The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. J. Exerc. Nutr. Biochem. 2017, 21, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef]
- Thornalley, P.J. Dicarbonyl intermediates in the maillard reaction. Ann. N. Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef]
- Alavi, P.; Yousefi, R.; Amirghofran, S.; Karbalaei-Heidari, H.R.; Moosavi-Movahedi, A.A. Structural analysis and aggregation propensity of reduced and nonreduced glycated insulin adducts. Appl. Biochem. Biotechnol. 2013, 170, 623–638. [Google Scholar] [CrossRef]
- McNulty, M.; Mahmud, A.; Feely, J. Advanced glycation end-products and arterial stiffness in hypertension. Am. J. Hypertens. 2007, 20, 242–247. [Google Scholar] [CrossRef]
- Safar, M.E. Systolic blood pressure, pulse pressure and arterial stiffness as cardiovascular risk factors. Curr. Opin. Nephrol. Hypertens. 2001, 10, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Schram, M.T.; Schalkwijk, C.G.; Bootsma, A.H.; Fuller, J.H.; Chaturvedi, N.; Stehouwer, C.D. Advanced glycation end products are associated with pulse pressure in type 1 diabetes: The eurodiab prospective complications study. Hypertension 2005, 46, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Koh, Y.H.; Takahashi, M.; Miyamoto, Y.; Suzuki, K.; Dohmae, N.; Takio, K.; Honke, K.; Taniguchi, N. Identification of the binding site of methylglyoxal on glutathione peroxidase: Methylglyoxal inhibits glutathione peroxidase activity via binding to glutathione binding sites arg 184 and 185. Free Radic. Res. 2003, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Juurlink, B.H. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002, 39, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Desai, K.; Chang, T.; Wu, L. Vascular methylglyoxal metabolism and the development of hypertension. J. Hypertens. 2005, 23, 1565–1573. [Google Scholar] [CrossRef]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arter. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef]
- Vasdev, S.; Gill, V.; Singal, P. Role of advanced glycation end products in hypertension and atherosclerosis: Therapeutic implications. Cell Biochem. Biophys. 2007, 49, 48–63. [Google Scholar]
- Cheng, C.L.; Tang, Y.; Zheng, Z.; Liu, X.; Ye, Z.C.; Wang, C.; Lou, T.Q. Advanced glycation end-products activate the renin-angiotensin system through the rage/pi3-k signaling pathway in podocytes. Clin. Investig. Med. 2012, 35, 18701. [Google Scholar] [CrossRef]
- Haimoto, H.; Sasakabe, T.; Wakai, K.; Umegaki, H. Effects of a low-carbohydrate diet on glycemic control in outpatients with severe type 2 diabetes. Nutr. Metab. 2009, 6, 1743–7075. [Google Scholar] [CrossRef]
- Kaburagi, T.; Kanaki, K.; Otsuka, Y.; Hino, R. Low-carbohydrate diet inhibits different advanced glycation end products in kidney depending on lipid composition but causes adverse morphological changes in a non-obese model mice. Nutrients 2019, 11, 2801. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Zavala-Flores, L.; Anandhan, A.; Wang, F.; Skotak, M.; Chandra, N.; Li, M.; Pappa, A.; Martinez-Fong, D.; Del Razo, L.M.; et al. Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 2014, 142, 206–230. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. Elastin in large artery stiffness and hypertension. J. Cardiovasc. Transl. Res. 2012, 5, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, C.A.; Tharaux, P.L.; Lehoux, S. Extracellular matrix alterations in hypertensive vascular remodeling. J. Mol. Cell. Cardiol. 2010, 48, 433–439. [Google Scholar] [CrossRef]
- Elia, A.; Fossati, S. Autonomic nervous system and cardiac neuro-signaling pathway modulation in cardiovascular disorders and alzheimer’s disease. Front. Physiol. 2023, 14, 1060666. [Google Scholar] [CrossRef]
- Belo, V.A.; Guimarães, D.A.; Castro, M.M. Matrix metalloproteinase 2 as a potential mediator of vascular smooth muscle cell migration and chronic vascular remodeling in hypertension. J. Vasc. Res. 2015, 52, 221–231. [Google Scholar] [CrossRef]
- Massaro, M.; Scoditti, E.; Carluccio, M.A.; De Caterina, R. Oxidative stress and vascular stiffness in hypertension: A renewed interest for antioxidant therapies? Vasc. Pharmacol. 2019, 116, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ros). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Tian, W.; Wei, T.; Liu, F. The neuroprotective effects of β-hydroxybutyrate on aβ-injected rat hippocampus in vivo and in aβ-treated pc-12 cells in vitro. Free Radic. Res. 2015, 49, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bersch-Ferreira, Â.C.; Sampaio, G.R.; Gehringer, M.O.; da Silva Torres, E.A.F.; Ross-Fernandes, M.B.; da Silva, J.T.; Torreglosa, C.R.; Kovacs, C.; Alves, R.; Magnoni, C.D.; et al. Association between plasma fatty acids and inflammatory markers in patients with and without insulin resistance and in secondary prevention of cardiovascular disease, a cross-sectional study. Nutr. J. 2018, 17, 26. [Google Scholar] [CrossRef]
- Loperena, R.; Harrison, D.G. Oxidative stress and hypertensive diseases. Med. Clin. 2017, 101, 169–193. [Google Scholar] [CrossRef]
- Loperena, R.; Van Beusecum, J.P.; Itani, H.A.; Engel, N.; Laroumanie, F.; Xiao, L.; Elijovich, F.; Laffer, C.L.; Gnecco, J.S.; Noonan, J.; et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of stat3, interleukin 6 and hydrogen peroxide. Cardiovasc. Res. 2018, 114, 1547–1563. [Google Scholar] [CrossRef] [PubMed]
- Henegar, J.R.; Bigler, S.A.; Henegar, L.K.; Tyagi, S.C.; Hall, J.E. Functional and structural changes in the kidney in the early stages of obesity. J. Am. Soc. Nephrol. 2001, 12, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation 2016, 133, 894–906. [Google Scholar] [CrossRef]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr. Atheroscler. Rep. 2017, 19, 017–0691. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Koutlas, V.; Pappas, C.; Mitsis, M.; Dounousi, E. The endothelial glycocalyx as a target of ischemia and reperfusion injury in kidney transplantation-where have we gone so far? Int. J. Mol. Sci. 2021, 22, 2157. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M. Endothelial glycocalyx damage as a systemic inflammatory microvascular endotheliopathy in COVID-19. Biomed. J. 2020, 43, 399–413. [Google Scholar] [CrossRef]
- Noble, M.I.; Drake-Holland, A.J.; Vink, H. Hypothesis: Arterial glycocalyx dysfunction is the first step in the atherothrombotic process. Qjm 2008, 101, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Bäck, M.; Bochaton-Piallat, M.L.; Caligiuri, G.; Daemen, M.J.; Davies, P.F.; Hoefer, I.E.; Holvoet, P.; Jo, H.; Krams, R.; et al. Biomechanical factors in atherosclerosis: Mechanisms and clinical implications. Eur. Heart J. 2014, 35, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, ldl deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Dabagh, M.; Jalali, P.; Tarbell, J.M. The transport of ldl across the deformable arterial wall: The effect of endothelial cell turnover and intimal deformation under hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H983–H996. [Google Scholar]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of ldl by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High density lipoprotein cholesterol efflux capacity and atherosclerosis in cardiovascular disease: Pathophysiological aspects and pharmacological perspectives. Cells 2021, 10, 574. [Google Scholar] [CrossRef]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.; Curatolo, N.; Benoist, J.F.; Auvin, S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015, 56, e95–e98. [Google Scholar] [CrossRef]
- Alnami, A.; Bima, A.; Alamoudi, A.; Eldakhakhny, B.; Sakr, H.; Elsamanoudy, A. Modulation of dyslipidemia markers apo b/apo a and triglycerides/hdl-cholesterol ratios by low-carbohydrate high-fat diet in a rat model of metabolic syndrome. Nutrients 2022, 14, 1903. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of oxldl and its impact on cardiovascular health: Focus on atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Vrieling, F.; Wilson, L.; Rensen, P.C.N.; Walzl, G.; Ottenhoff, T.H.M.; Joosten, S.A. Oxidized low-density lipoprotein (oxLDL) supports mycobacterium tuberculosis survival in macrophages by inducing lysosomal dysfunction. PLoS Pathog. 2019, 15, e1007724. [Google Scholar] [CrossRef]
- Patel, V.I.; Patel, K.P.; Makadia, M.G.; Shah, A.D.; Chaudhari, K.S.; Nilayangode, H.N. Levels of apolipoprotein a1, b100 and lipoprotein (a) in controlled and uncontrolled diabetic patients and in non-diabetic healthy people. J. Clin. Diagn. Res. 2017, 11, BC01. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yang, Y.; Zhang, J.; Wang, Y.; He, W.; Zhang, X.; Zhu, J.; Lu, Z. The apob100/apoai ratio is independently associated with the severity of coronary heart disease: A cross sectional study in patients undergoing coronary angiography. Lipids Health Dis. 2015, 14, 150. [Google Scholar] [CrossRef]
- Gagliardino, J.J.; Salazar, M.R.; Espeche, W.G.; Tolosa Chapasian, P.E.; Gomez Garizoain, D.; Olano, R.D.; Stavile, R.N.; Balbín, E.; Martinez, C.; Leiva Sisnieguez, B.C.; et al. Arterial stiffness: Its relation with prediabetes and metabolic syndrome and possible pathogenesis. J. Clin. Med. 2021, 10, 3251. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ah Lee, Y.; Yong Lee, S.; Ho Shin, C.; Hyun Kim, J. Comparison of lipid-derived markers for metabolic syndrome in youth: Triglyceride/hdl cholesterol ratio, triglyceride-glucose index, and non-hdl cholesterol. Tohoku J. Exp. Med. 2022, 256, 53–62. [Google Scholar] [CrossRef]
- Aslan Çin, N.N.; Yardımcı, H.; Koç, N.; Uçaktürk, S.A.; Akçil Ok, M. Triglycerides/high-density lipoprotein cholesterol is a predictor similar to the triglyceride-glucose index for the diagnosis of metabolic syndrome using international diabetes federation criteria of insulin resistance in obese adolescents: A cross-sectional study. J. Pediatr. Endocrinol. Metab. 2020, 33, 777–784. [Google Scholar]
- Buga, A.; Welton, G.L.; Scott, K.E.; Atwell, A.D.; Haley, S.J.; Esbenshade, N.J.; Abraham, J.; Buxton, J.D.; Ault, D.L.; Raabe, A.S.; et al. The effects of carbohydrate versus fat restriction on lipid profiles in highly trained, recreational distance runners: A randomized, cross-over trial. Nutrients 2022, 14, 1135. [Google Scholar] [CrossRef]
- Okuda, T. A low-carbohydrate ketogenic diet induces the expression of very-low-density lipoprotein receptor in liver and affects its associated metabolic abnormalities. Npj Sci. Food 2019, 3, 25. [Google Scholar] [CrossRef]
- Larsen, J.B.; Hvas, A.M. Fibrin clot formation and lysis in plasma. Methods Protoc. 2020, 3, 67. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin formation, structure and properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.C.; Nosalski, R.; MacRitchie, N.; Tomaszewski, M.; Maffia, P.; Harrison, D.G.; Guzik, T.J. Therapeutic targeting of inflammation in hypertension: From novel mechanisms to translational perspective. Cardiovasc. Res. 2021, 117, 2589–2609. [Google Scholar] [CrossRef]
- Pan, S. Molecular mechanisms responsible for the atheroprotective effects of laminar shear stress. Antioxid. Redox Signal. 2009, 11, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Ozkor, M.A.; Quyyumi, A.A. Endothelium-derived hyperpolarizing factor and vascular function. Cardiol. Res. Pract. 2011, 2011, 156146. [Google Scholar] [CrossRef]
- He, M.; Martin, M.; Marin, T.; Chen, Z.; Gongol, B. Endothelial mechanobiology. APL Bioeng. 2020, 4, 010904. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Berk, B.C. Novel mechanisms of endothelial mechanotransduction. Arter. Thromb. Vasc. Biol. 2014, 34, 2378–2386. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Chabowski, D.S.; Kadlec, A.O.; Durand, M.J.; Freed, J.K.; Ait-Aissa, K.; Beyer, A.M. The human microcirculation: Regulation of flow and beyond. Circ. Res. 2016, 118, 157–172. [Google Scholar] [CrossRef]
- Bourdeau, J.E.; Chen, E.R.; Carone, F.A. Insulin uptake in the renal proximal tubule. Am. J. Physiol. 1973, 225, 1399–1404. [Google Scholar] [CrossRef]
- Nakamura, R.; Emmanouel, D.S.; Katz, A.I. Insulin binding sites in various segments of the rabbit nephron. J. Clin. Investig. 1983, 72, 388–392. [Google Scholar] [CrossRef]
- Butlen, D.; Vadrot, S.; Roseau, S.; Morel, F. Insulin receptors along the rat nephron: [125i] insulin binding in microdissected glomeruli and tubules. Pflug. Arch. 1988, 412, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Gesek, F.A.; Schoolwerth, A.C. Insulin increases Na(+)-H+ exchange activity in proximal tubules from normotensive and hypertensive rats. Am. J. Physiol. 1991, 260, F695–F703. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kwon, W.; Kawano, K.; Choi, J.W.; Kim, J.H.; Donowitz, M. Lysophosphatidic acid stimulates brush border Na+/H+ exchanger 3 (NHE3) activity by increasing its exocytosis by an NHE3 kinase a regulatory protein-dependent mechanism. J. Biol. Chem. 2003, 278, 16494–16501. [Google Scholar] [CrossRef] [PubMed]
- Shiue, H.; Musch, M.W.; Wang, Y.; Chang, E.B.; Turner, J.R. Akt2 phosphorylates ezrin to trigger nhe3 translocation and activation. J. Biol. Chem. 2005, 280, 1688–1695. [Google Scholar] [CrossRef]
- Baum, M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J. Clin. Investig. 1987, 79, 1104–1109. [Google Scholar] [CrossRef]
- Klisic, J.; Hu, M.C.; Nief, V.; Reyes, L.; Fuster, D.; Moe, O.W.; Ambühl, P.M. Insulin activates Na(+)/H(+) exchanger 3: Biphasic response and glucocorticoid dependence. Am. J. Physiol. Ren. Physiol. 2002, 283, F532–A539. [Google Scholar] [CrossRef]
- Fuster, D.G.; Bobulescu, I.A.; Zhang, J.; Wade, J.; Moe, O.W. Characterization of the regulation of renal Na+/H+ exchanger nhe3 by insulin. Am. J. Physiol. Ren. Physiol. 2007, 292, F577–F585. [Google Scholar] [CrossRef]
- Féraille, E.; Carranza, M.L.; Gonin, S.; Béguin, P.; Pedemonte, C.; Rousselot, M.; Caverzasio, J.; Geering, K.; Martin, P.Y.; Favre, H. Insulin-induced stimulation of Na+,K+-atpase activity in kidney proximal tubule cells depends on phosphorylation of the alpha-subunit at tyr-10. Mol. Biol. Cell 1999, 10, 2847–2859. [Google Scholar] [CrossRef]
- Rivera, C.; Santos-Reyes, H.; Martinez-Maldonado, M. Response of dog renal Na+, K+-atpase to insulin in vitro. Kidney Blood Press. Res. 1978, 1, 74–83. [Google Scholar] [CrossRef]
- Feraille, E.; Carranza, M.L.; Rousselot, M.; Favre, H. Insulin enhances sodium sensitivity of na-k-atpase in isolated rat proximal convoluted tubule. Am. J. Physiol.—Ren. Physiol. 1994, 267, F55–F62. [Google Scholar] [CrossRef]
- Ruiz, O.S.; Qiu, Y.-Y.; Cardoso, L.R.; Arruda, J.A. Regulation of the renal na–hco3 cotransporter: Ix. Modulation by insulin, epidermal growth factor and carbachol. Regul. Pept. 1998, 77, 155–161. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Maron, R.; Kahn, C.R. Insulin rapidly stimulates tyrosine phosphorylation of a m r-185,000 protein in intact cells. Nature 1985, 318, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate irs-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Wang, L.-M.; Zhang, Y.; Yenush, L.; Myers Jr, M.G.; Glasheen, E.; Lane, W.S.; Pierce, J.H.; White, M.F. Role of irs-2 in insulin and cytokine signalling. Nature 1995, 377, 173–177. [Google Scholar] [CrossRef]
- Brüning, J.C.; Winnay, J.; Cheatham, B.; Kahn, C.R. Differential signaling by insulin receptor substrate 1 (irs-1) and irs-2 in irs-1-deficient cells. Mol. Cell. Biol. 1997, 17, 1513–1521. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.-E.; Brüning, J.C.; Haag III, B.; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the irs-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef]
- Kubota, N.; Tobe, K.; Terauchi, Y.; Eto, K.; Yamauchi, T.; Suzuki, R.; Tsubamoto, Y.; Komeda, K.; Nakano, R.; Miki, H. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 2000, 49, 1880–1889. [Google Scholar] [CrossRef]
- Kido, Y.; Burks, D.J.; Withers, D.; Bruning, J.C.; Kahn, C.R.; White, M.F.; Accili, D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, irs-1, and irs-2. J. Clin. Investig. 2000, 105, 199–205. [Google Scholar] [CrossRef]
- Nakamura, M.; Yamazaki, O.; Shirai, A.; Horita, S.; Satoh, N.; Suzuki, M.; Hamasaki, Y.; Noiri, E.; Kume, H.; Enomoto, Y.; et al. Preserved Na/HCO3 cotransporter sensitivity to insulin may promote hypertension in metabolic syndrome. Kidney Int. 2015, 87, 535–542. [Google Scholar] [CrossRef]
- Horita, S.; Seki, G.; Yamada, H.; Suzuki, M.; Koike, K.; Fujita, T. Insulin resistance, obesity, hypertension, and renal sodium transport. Int. J. Hypertens. 2011, 2011, 391762. [Google Scholar] [CrossRef]
- Goodyear, L.J.; Giorgino, F.; Sherman, L.A.; Carey, J.; Smith, R.J.; Dohm, G.L. Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J. Clin. Investig. 1995, 95, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Ishizuka, T.; Shao, J.; Huston, L.; Highman, T.; Catalano, P. Impaired glucose transport and insulin receptor tyrosine phosphorylation in skeletal muscle from obese women with gestational diabetes. Diabetes 1999, 48, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Rondinone, C.M.; Wang, L.-M.; Lonnroth, P.; Wesslau, C.; Pierce, J.H.; Smith, U. Insulin receptor substrate (irs) 1 is reduced and irs-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA 1997, 94, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, E.; Jansson, P.A.; Axelsen, M.; Eriksson, J.W.; Huang, X.; Groop, L.; Rondinone, C.; Sjöström, L.; Smith, U. Low cellular irs 1 gene and protein expression predict insulin resistance and niddm. FASEB J. 1999, 13, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Unwin, D.J.; Tobin, S.D.; Murray, S.W.; Delon, C.; Brady, A.J. Substantial and sustained improvements in blood pressure, weight and lipid profiles from a carbohydrate restricted diet: An observational study of insulin resistant patients in primary care. Int. J. Environ. Res. Public Health 2019, 16, 2680. [Google Scholar] [CrossRef] [PubMed]
- Unwin, D.; Unwin, J. Low carbohydrate diet to achieve weight loss and improve hba1c in type 2 diabetes and pre-diabetes: Experience from one general practice. Pract. Diabetes 2014, 31, 76–79. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakr, H.F.; Sirasanagandla, S.R.; Das, S.; Bima, A.I.; Elsamanoudy, A.Z. Insulin Resistance and Hypertension: Mechanisms Involved and Modifying Factors for Effective Glucose Control. Biomedicines 2023, 11, 2271. https://doi.org/10.3390/biomedicines11082271
Sakr HF, Sirasanagandla SR, Das S, Bima AI, Elsamanoudy AZ. Insulin Resistance and Hypertension: Mechanisms Involved and Modifying Factors for Effective Glucose Control. Biomedicines. 2023; 11(8):2271. https://doi.org/10.3390/biomedicines11082271
Chicago/Turabian StyleSakr, Hussein F., Srinivasa Rao Sirasanagandla, Srijit Das, Abdulhadi I. Bima, and Ayman Z. Elsamanoudy. 2023. "Insulin Resistance and Hypertension: Mechanisms Involved and Modifying Factors for Effective Glucose Control" Biomedicines 11, no. 8: 2271. https://doi.org/10.3390/biomedicines11082271
APA StyleSakr, H. F., Sirasanagandla, S. R., Das, S., Bima, A. I., & Elsamanoudy, A. Z. (2023). Insulin Resistance and Hypertension: Mechanisms Involved and Modifying Factors for Effective Glucose Control. Biomedicines, 11(8), 2271. https://doi.org/10.3390/biomedicines11082271