
Unlocking the Molecular Secrets of Antifolate Drug Resistance: A Multi-Omics Investigation of the NCI-60 Cell Line Panel


Abstract
:1. Introduction
2. Materials and Methods
2.1. Procurement of Publicly Available Omics Datasets of the NCI-60 Cell Line Panel
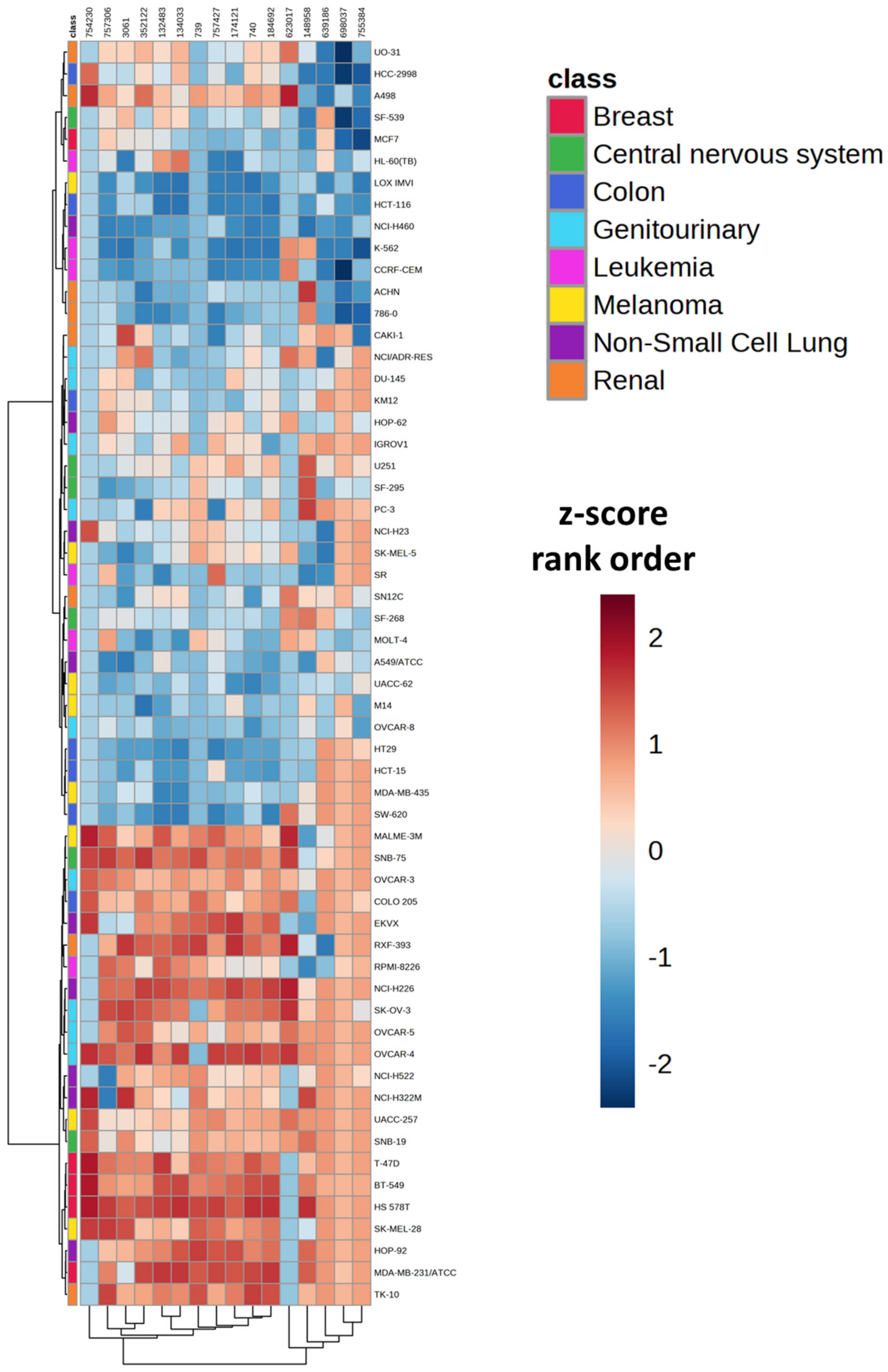
2.2. Assessment of Antifolate Drug Response and Calculation of Overall Antifolate Sensitivity for the NCI-60 Cell Line Panel
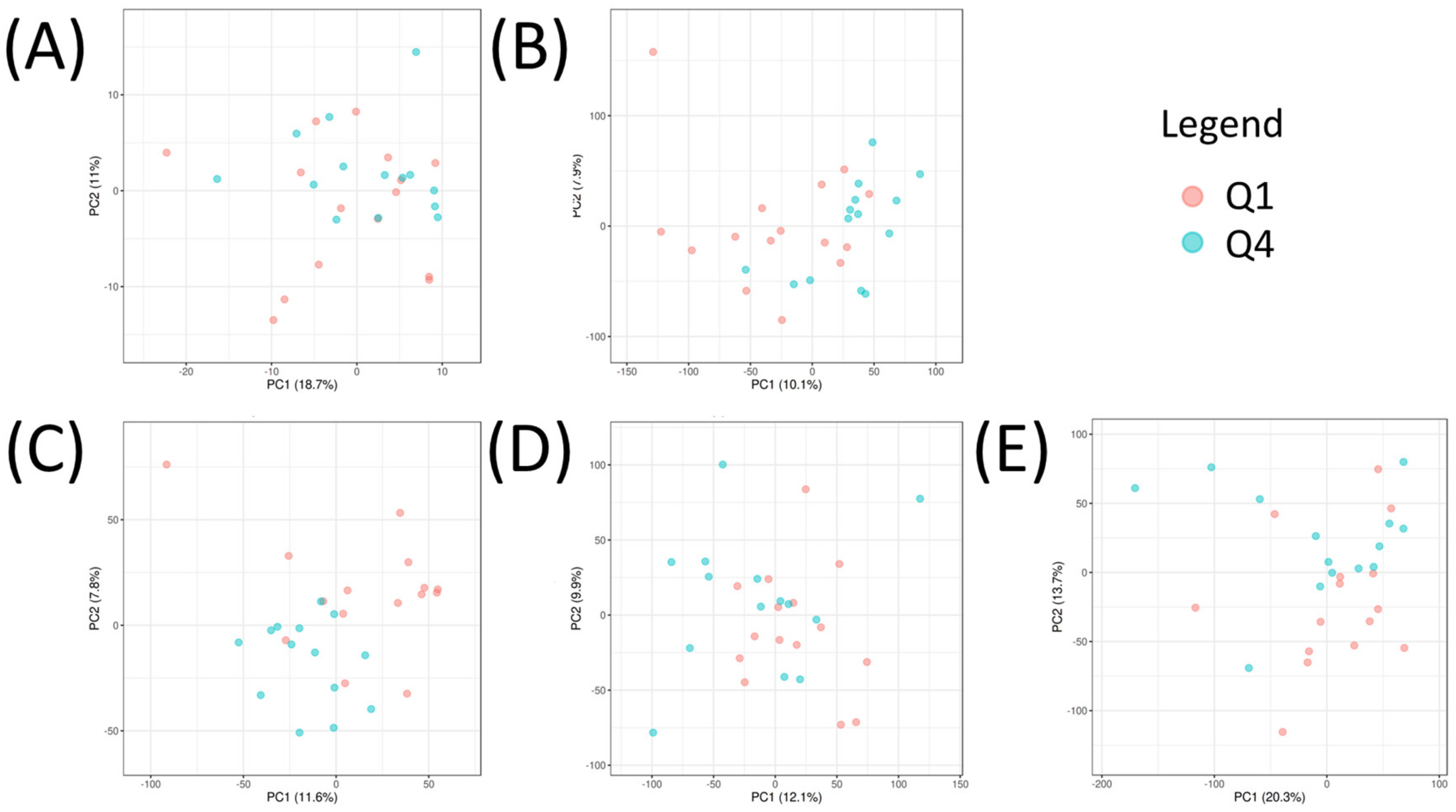
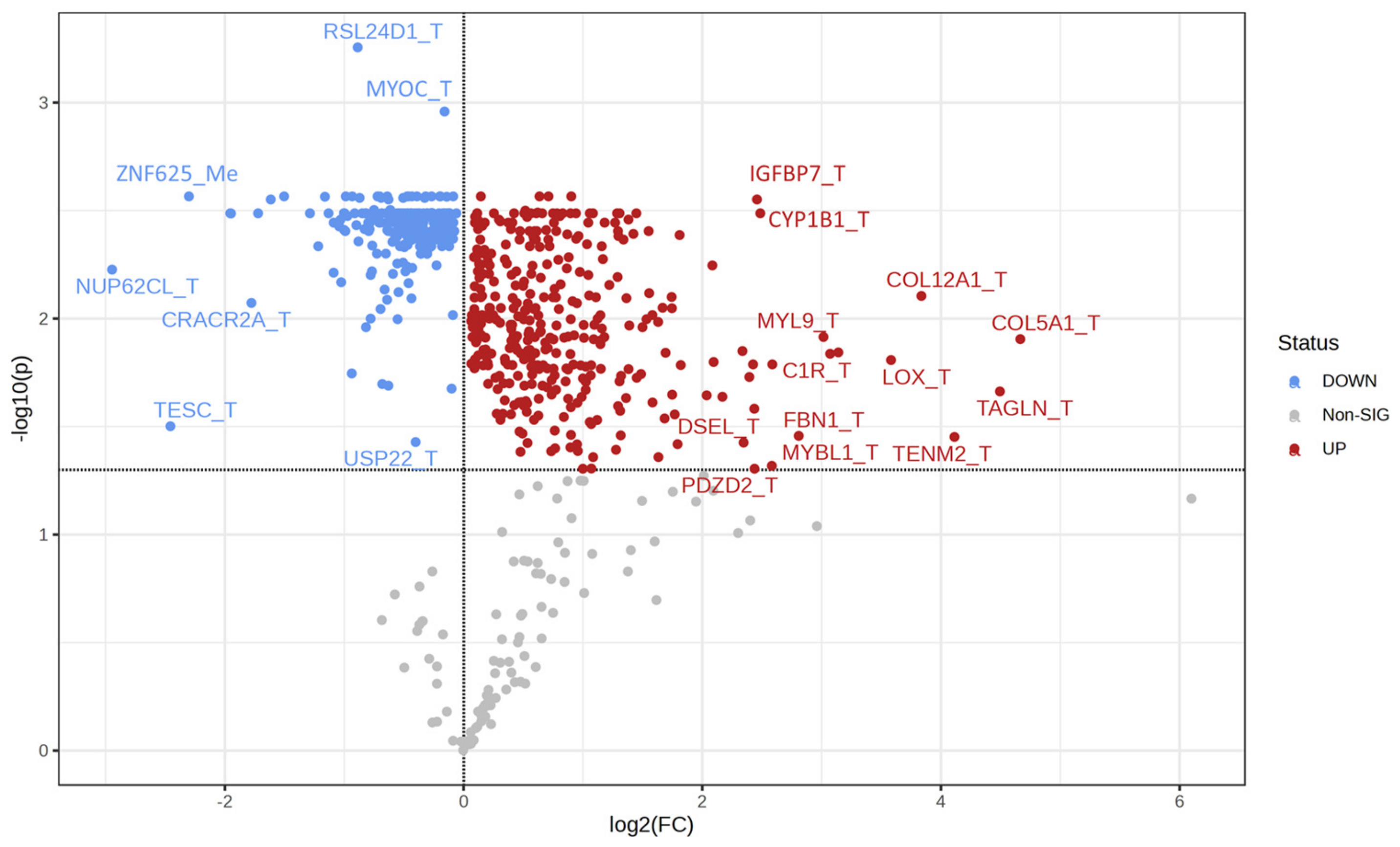
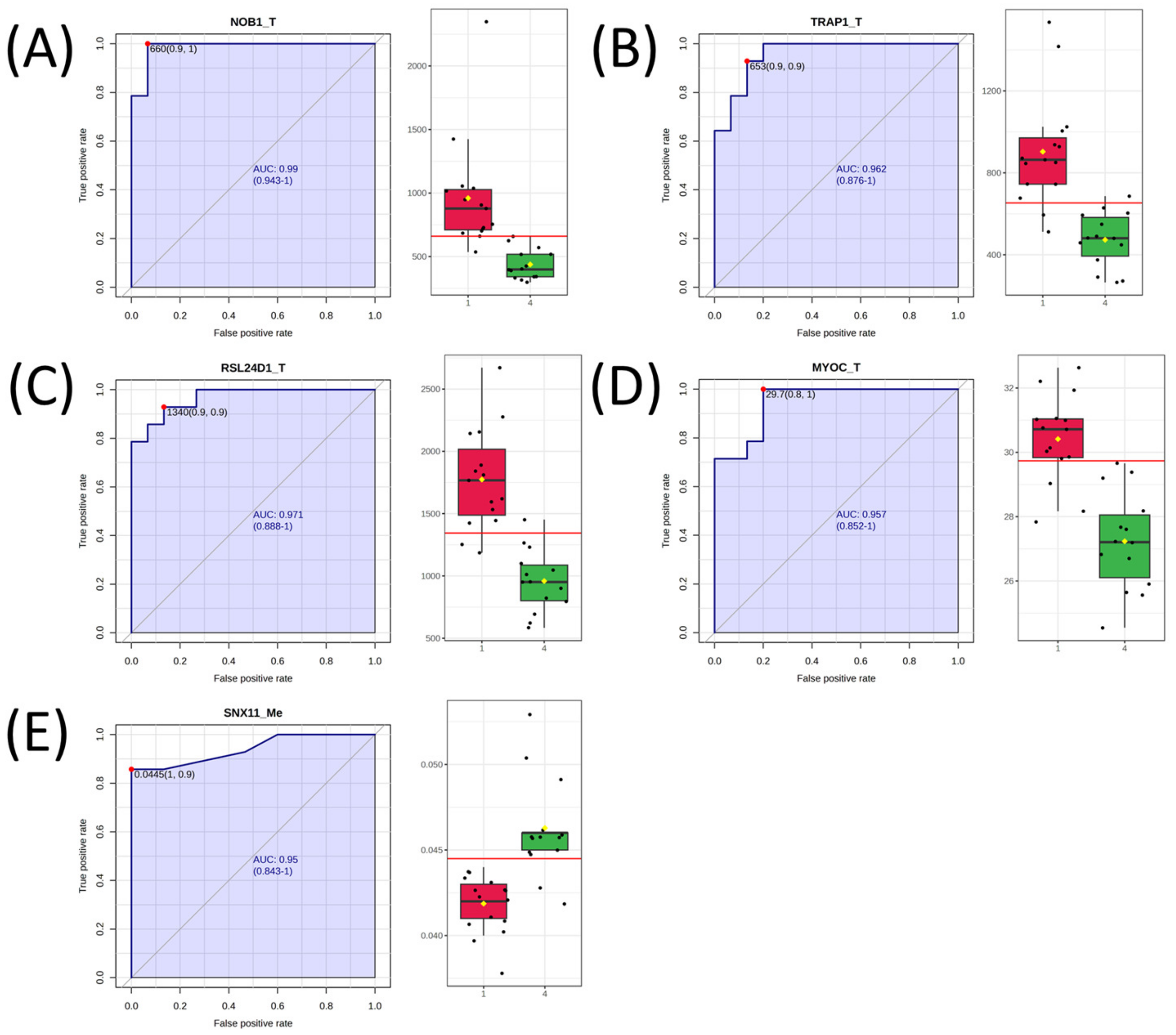
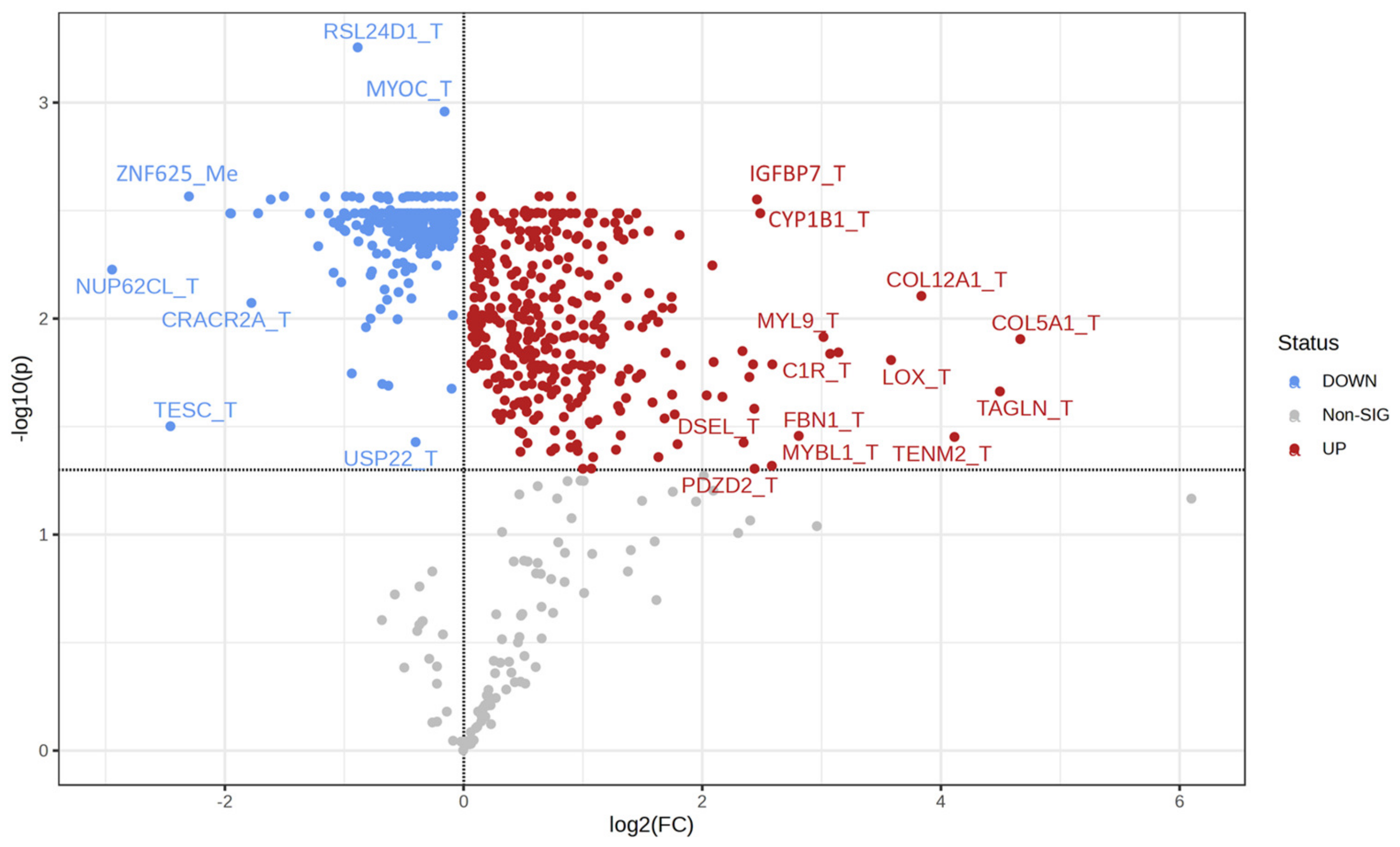
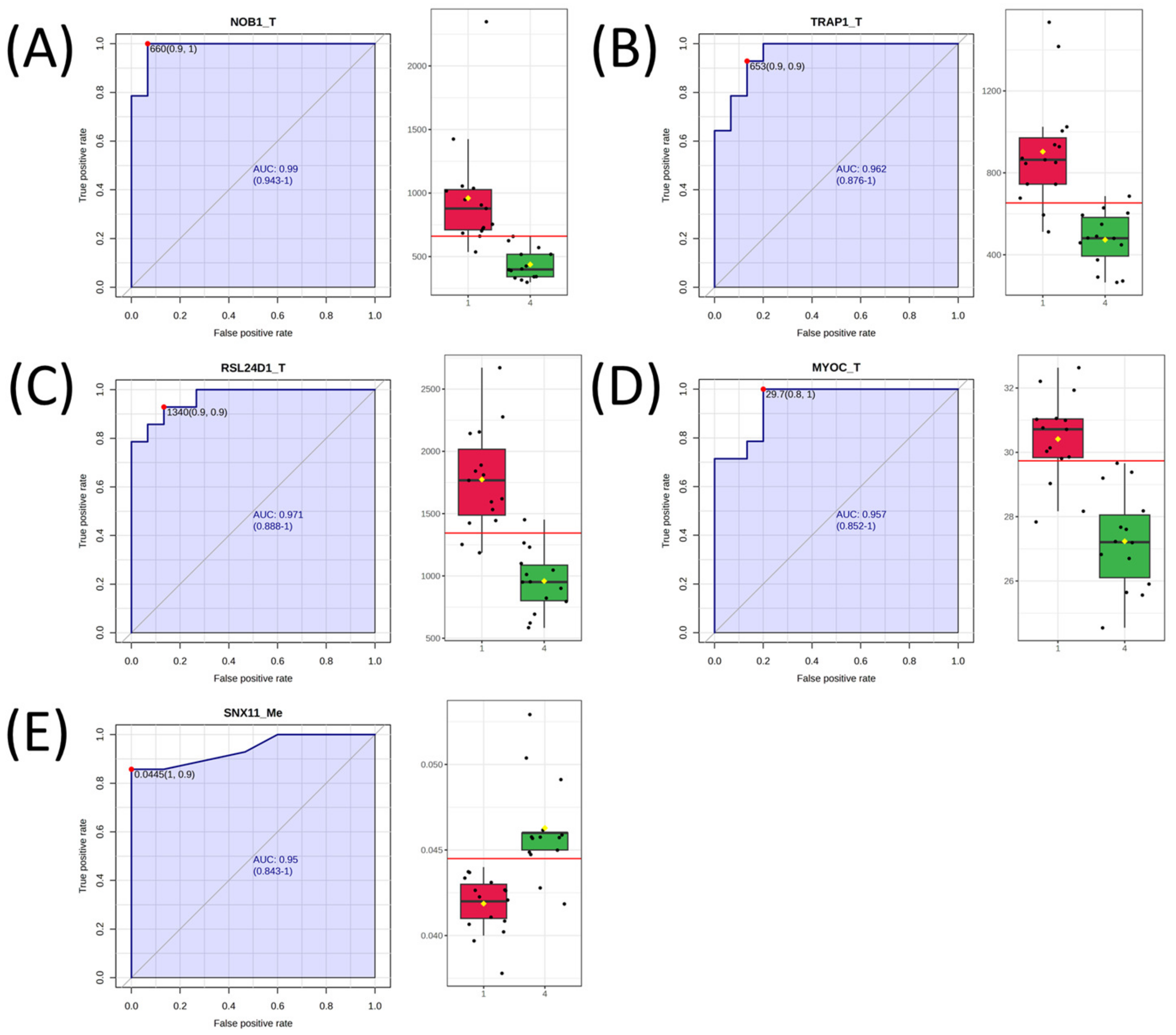
2.3. Univariate Analysis and Network Analysis of Features Differentiating Cell Lines Sensitive or Resistant to Antifolates
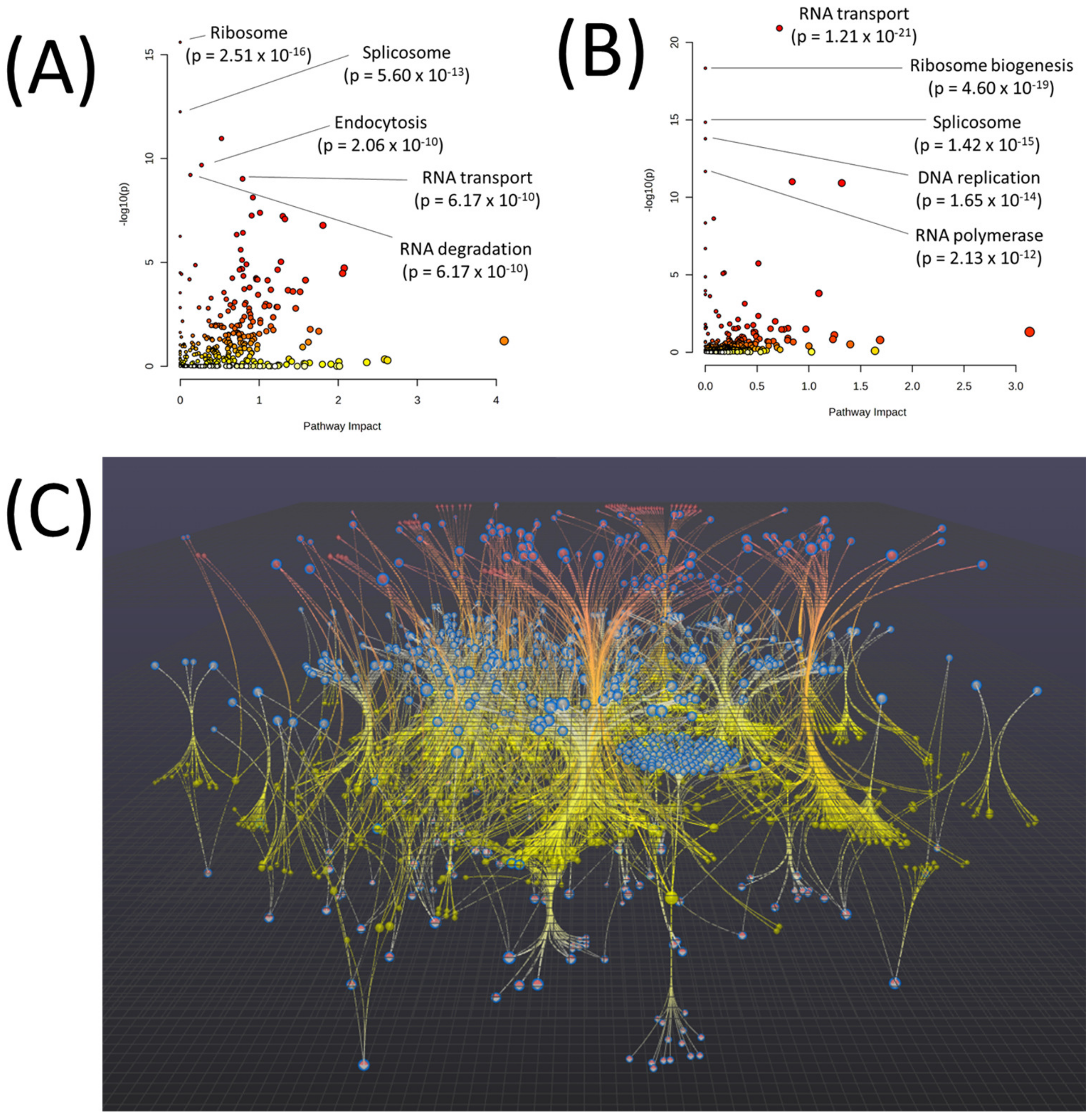
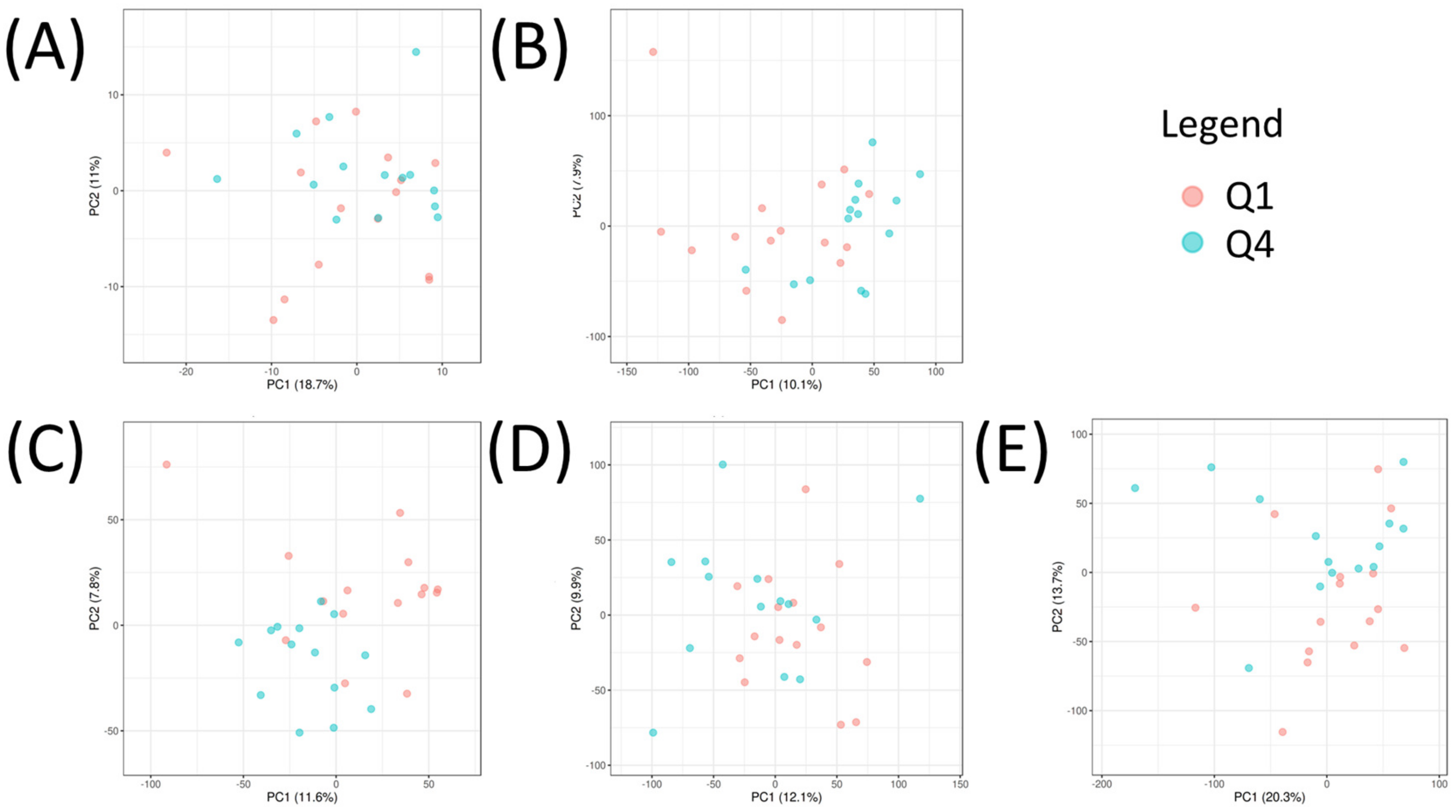
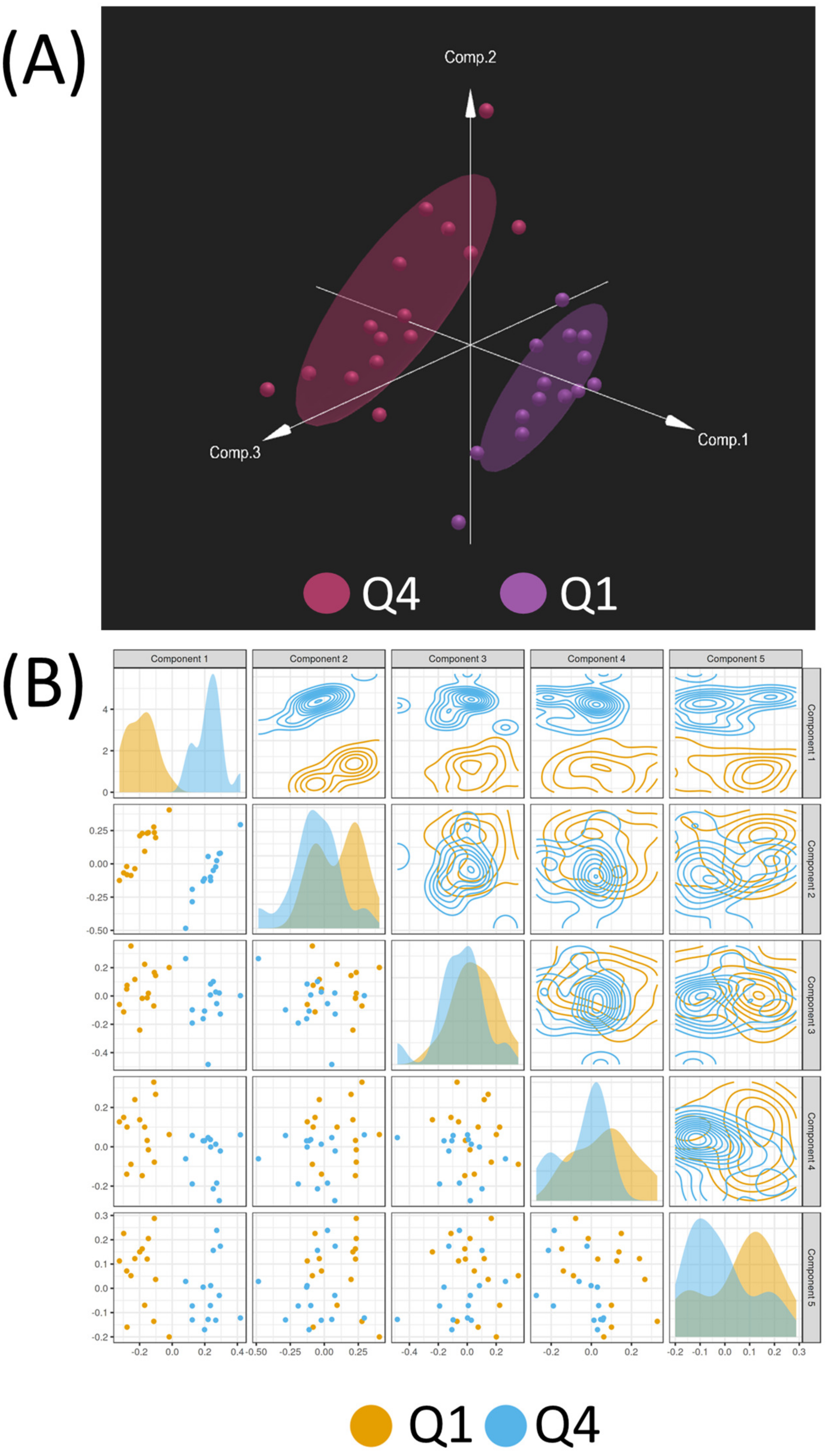
2.4. Multi-Omic Multivariate Analysis and Joint Pathway Analysis of Sensitive versus Resistant Cell Lines to Antifolates
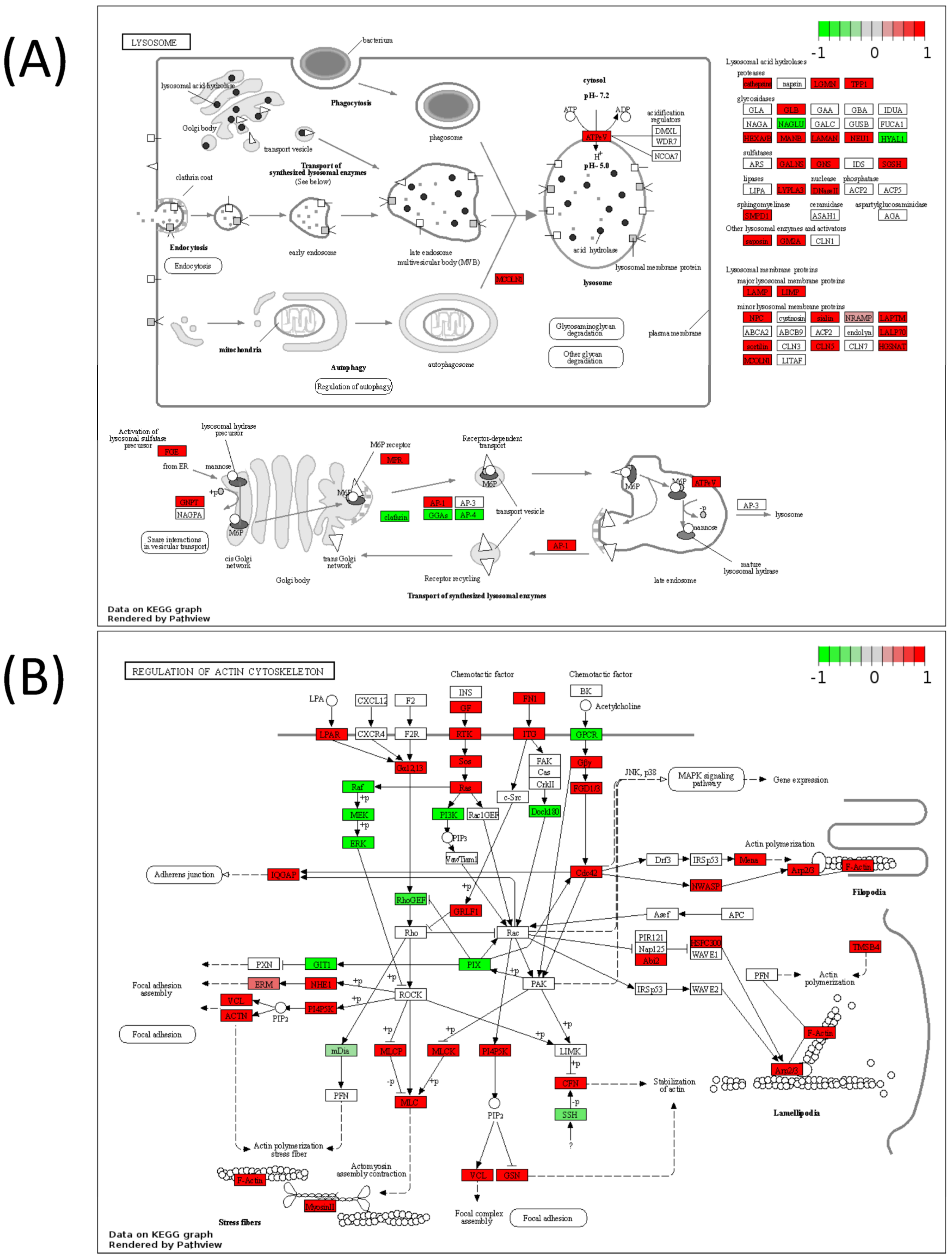
2.5. Pathview Analysis for Integrative Pathway Visualization
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-jones, S.; Zhou, X.; Cai, S.; Tu, Y.; Mccoy, A.; Peoples, M.; et al. Resistance to neoadjuvant chemotherapy in triple negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019, 11, eaav0936. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, D.C.; Cheng, Y.; Qian, M.; Zhang, M.; Shen, Q.; Wang, X. Roles of tumor heterogeneity in the development of drug resistance: A call for precision therapy. Semin. Cancer Biol. 2017, 42, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Visentin, M.; Zhao, R.; Goldman, I.D. The Antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef]
- Gonen, N.; Assaraf, Y.G. Antifolates in cancer therapy: Structure, activity and mechanisms of drug resistance. Drug Resist. Updat. 2012, 15, 183–210. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updat. 2020, 53, 100715. [Google Scholar] [CrossRef]
- Jung, H.D.; Sung, Y.J.; Kim, H.U. Omics and Computational Modeling Approaches for the Effective Treatment of Drug-Resistant Cancer Cells. Front. Genet. 2021, 12, 742902. [Google Scholar] [CrossRef]
- Celebi, R.; Bear Don’t Walk, O., IV; Movva, R.; Alpsoy, S.; Dumontier, M. In-Silico Prediction of Synergistic Anti-Cancer Drug Combinations Using Multi-omics Data. Sci. Rep. 2019, 9, 8949. [Google Scholar] [CrossRef]
- Chabner, B.A. NCI-60 Cell Line Screening: A Radical Departure in Its Time. J. Natl. Cancer Inst. 2016, 108, djv388. [Google Scholar] [CrossRef] [PubMed]
- Kohn, K.W.; Zeeberg, B.M.; Reinhold, W.C.; Pommier, Y. Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS ONE 2014, 9, e99269. [Google Scholar] [CrossRef] [PubMed]
- Pfister, T.D.; Reinhold, W.C.; Agama, K.; Gupta, S.; Khin, S.A.; Kinders, R.J.; Parchment, R.E.; Tomaszewski, J.E.; Doroshow, J.H.; Pommier, Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 2009, 8, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Frejno, M.; Meng, C.; Ruprecht, B.; Oellerich, T.; Scheich, S.; Kleigrewe, K.; Drecoll, E.; Samaras, P.; Hogrebe, A.; Helm, D.; et al. Proteome activity landscapes of tumor cell lines determine drug responses. Nat. Commun. 2020, 11, 3639. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Shankavaram, U.T.; Varma, S.; Kane, D.; Sunshine, M.; Chary, K.K.; Reinhold, W.C.; Pommier, Y.; Weinstein, J.N. CellMiner: A relational database and query tool for the NCI-60 cancer cell lines. BMC Genom. 2009, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, 388–396. [Google Scholar] [CrossRef]
- Zhou, G.; Pang, Z.; Lu, Y.; Ewald, J.; Xia, J. OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022, 50, W527–W533. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Zhou, G.; Ewald, J.; Xia, J. OmicsAnalyst: A comprehensive web-based platform for visual analytics of multi-omics data. Nucleic Acids Res. 2021, 49, W476–W482. [Google Scholar] [CrossRef]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Cao, K.A.L. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef] [PubMed]
- Tibor Fekete, J.; Győrffy, B. A unified platform enabling biomarker ranking and validation for 1562 drugs using transcriptomic data of 1250 cancer cell lines. Comput. Struct. Biotechnol. J. 2022, 20, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R. Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents. Int. J. Mol. Sci. 2023, 24, 13242. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Molina, S.; Sumner, S. Metabolomics Analysis Reveals Altered Metabolic Pathways and Response to Doxorubicin in Drug-Resistant Triple-Negative Breast Cancer Cells. Metabolites 2023, 13, 865. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Wiggs, A.; Molina, S.; Schroder, M.; Sumner, S. Metabolomics Analysis Reveals Novel Targets of Chemosensitizing Polyphenols and Omega-3 Polyunsaturated Fatty Acids in Triple Negative Breast Cancer Cells. Int. J. Mol. Med. 2023, 24, 4406. [Google Scholar] [CrossRef]
- Hohmann, T.; Dehghani, F. The cytoskeleton—A complex interacting meshwork. Cells 2019, 8, 362. [Google Scholar] [CrossRef]
- Kruppa, A.J.; Buss, F. Motor proteins at the mitochondria–cytoskeleton interface. J. Cell Sci. 2021, 134, jcs226084. [Google Scholar] [CrossRef]
- Pollard, T.D.; Goldman, R.D. Overview of the cytoskeleton from an evolutionary perspective. Cold Spring Harb. Perspect. Biol. 2018, 10, a030288. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The Cytoskeleton–Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Kosach, V.R.; Cherednyk, O.V.; Khoruzhenko, A.I. Characteristic of mTOR signaling and its involvement in the regulation of cell movements through remodeling the cytoskeleton architecture. Biopolym. Cell 2015, 31, 5–14. [Google Scholar] [CrossRef]
- Malik, A.R.; Urbanska, M.; MacIas, M.; Skalecka, A.; Jaworski, J. Beyond control of protein translation: What we have learned about the non-canonical regulation and function of mammalian target of rapamycin (mTOR). Biochim. Biophys. Acta-Proteins Proteomics 2013, 1834, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.J.; Gorski, S.M. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia. Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Osaw, K. Role of Autophagy in Cancer. In Autophagy—A Double-Edged Sword—Cell Survival or Death? IntechOpen: London, UK, 2013; Volume 7, pp. 961–967. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta-Rev. Cancer 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Lin, C.J.; Tsao, Y.N.; Shu, C.W. Autophagy modulation as a potential targeted cancer therapy: From drug repurposing to new drug development. Kaohsiung J. Med. Sci. 2021, 37, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Berkers, C.R. The influence of metabolism on drug response in cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Kanehisa, M. Molecular network analysis of diseases and drugs in KEGG. Methods Mol. Biol. 2013, 939, 263–275. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cancer Type | Median z-Score Rank Order | Quartile Number |
|---|---|---|---|
| K-562 | Leukemia | 2 | 1 |
| LOX IMVI | Melanoma | 2.5 | 1 |
| HCT-116 | Colon | 4 | 1 |
| NCI-H460 | Non-Small Cell Lung | 4.5 | 1 |
| 786-0 | Renal | 5 | 1 |
| CCRF-CEM | Leukemia | 6 | 1 |
| SW-620 | Colon | 8.5 | 1 |
| HT29 | Colon | 9 | 1 |
| HCT-15 | Colon | 9.5 | 1 |
| ACHN | Renal | 12 | 1 |
| UACC-62 | Melanoma | 12.5 | 1 |
| A549/ATCC | Non-Small Cell Lung | 12.5 | 1 |
| MCF7 | Breast | 13 | 1 |
| OVCAR-8 | Genitourinary | 13 | 1 |
| M14 | Melanoma | 14 | 1 |
| MOLT-4 | Leukemia | 14.5 | 2 |
| HL-60(TB) | Leukemia | 15 | 2 |
| SR | Leukemia | 16 | 2 |
| MDA-MB-435 | Melanoma | 17 | 2 |
| SF-295 | Central nervous system | 18 | 2 |
| SF-539 | Central nervous system | 20.5 | 2 |
| CAKI-1 | Renal | 20.5 | 2 |
| SF-268 | Central nervous system | 21.5 | 2 |
| SN12C | Renal | 21.5 | 2 |
| HCC-2998 | Colon | 22 | 2 |
| DU-145 | Genitourinary | 22 | 2 |
| NCI/ADR-RES | Genitourinary | 23 | 2 |
| SK-MEL-5 | Melanoma | 24 | 2 |
| KM12 | Colon | 25.5 | 2 |
| HOP-62 | Non-Small Cell Lung | 25.5 | 2 |
| NCI-H23 | Non-Small Cell Lung | 26 | 3 |
| U251 | Central nervous system | 28.5 | 3 |
| IGROV1 | Genitourinary | 29 | 3 |
| PC-3 | Genitourinary | 29 | 3 |
| RPMI-8226 | Leukemia | 31 | 3 |
| UO-31 | Renal | 32 | 3 |
| NCI-H522 | Non-Small Cell Lung | 33 | 3 |
| SNB-19 | Central nervous system | 36.5 | 3 |
| NCI-H322M | Non-Small Cell Lung | 36.5 | 3 |
| OVCAR-5 | Genitourinary | 36.5 | 3 |
| UACC-257 | Melanoma | 37 | 3 |
| A498 | Renal | 38 | 3 |
| COLO 205 | Colon | 39.5 | 3 |
| OVCAR-3 | Genitourinary | 39.5 | 3 |
| SK-MEL-28 | Melanoma | 42 | 4 |
| MALME-3M | Melanoma | 42.5 | 4 |
| TK-10 | Renal | 43 | 4 |
| EKVX | Non-Small Cell Lung | 45.5 | 4 |
| SK-OV-3 | Genitourinary | 45.5 | 4 |
| T-47D | Breast | 47 | 4 |
| HOP-92 | Non-Small Cell Lung | 47 | 4 |
| RXF-393 | Renal | 47 | 4 |
| BT-549 | Breast | 48 | 4 |
| SNB-75 | Central nervous system | 48 | 4 |
| NCI-H226 | Non-Small Cell Lung | 49 | 4 |
| OVCAR-4 | Genitourinary | 49.5 | 4 |
| MDA-MB-231/ATCC | Breast | 53 | 4 |
| HS 578T | Breast | 54 | 4 |
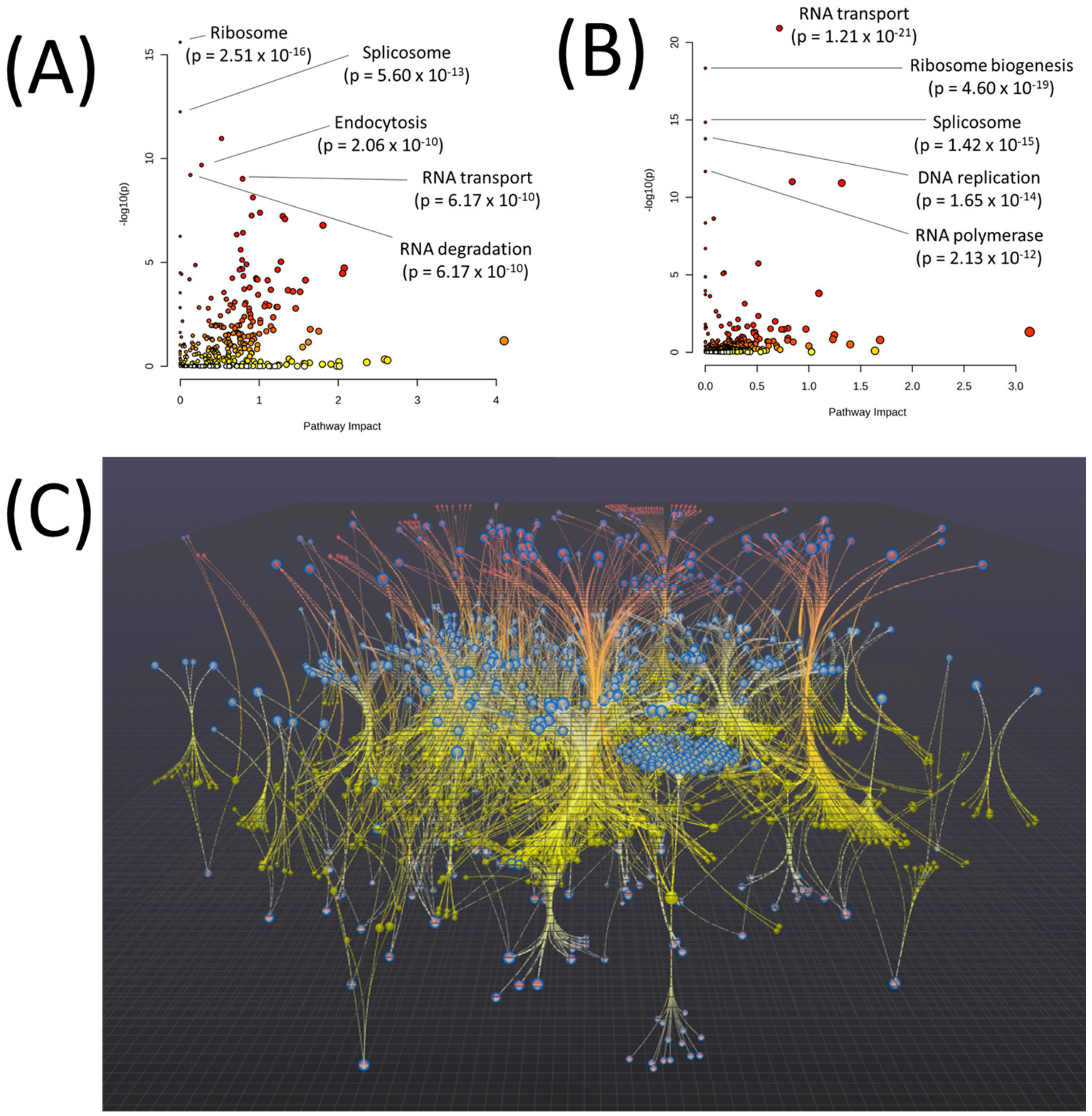
| Pathway | p-Value | FDR |
|---|---|---|
| EGFR tyrosine kinase inhibitor resistance | 1.03 × 10−165 | 3.45 × 10−163 |
| Glycolysis/Gluconeogenesis | 3.16 × 10−28 | 5.30 × 10−26 |
| Endocrine resistance | 1.84 × 10−24 | 2.06 × 10−22 |
| ABC transporters | 1.17 × 10−23 | 9.82 × 10−22 |
| Arginine and proline metabolism | 6.36 × 10−17 | 4.27 × 10−15 |
| Pyrimidine metabolism | 3.42 × 10−16 | 1.92 × 10−14 |
| beta-Alanine metabolism | 8.10 × 10−16 | 3.89 × 10−14 |
| Platinum drug resistance | 4.10 × 10−14 | 1.72 × 10−12 |
| Purine metabolism | 6.76 × 10−14 | 2.52 × 10−12 |
| Lysine degradation | 2.39 × 10−12 | 8.04 × 10−11 |
| Amino sugar and nucleotide sugar metabolism | 4.93 × 10−12 | 1.50 × 10−10 |
| Glutathione metabolism | 2.07 × 10−11 | 5.79 × 10−10 |
| Pentose phosphate pathway | 4.08 × 10−11 | 1.06 × 10−9 |
| Histidine metabolism | 6.18 × 10−11 | 1.48 × 10−9 |
| Fatty acid degradation | 6.89 × 10−11 | 1.54 × 10−9 |
| Gene Symbol | AUC | Log2 FC |
|---|---|---|
| NOB1_T | 0.986 | 1.13 |
| RSL24D1_T | 0.967 | 0.89 |
| TRAP1_T | 0.957 | 0.93 |
| MYOC_T | 0.948 | 0.16 |
| SNX11_Me | 0.940 | −0.14 |
| LOC101930306_T | 0.924 | 0.33 |
| STK26_T | 0.924 | 1.95 |
| ZNF500_T | 0.924 | 0.27 |
| DHODH_T | 0.919 | 0.75 |
| 234900_at_T | 0.914 | 0.20 |
| C10orf2_T | 0.914 | 0.82 |
| IGFBP7_T | 0.914 | −2.46 |
| DDX51_T | 0.910 | 0.65 |
| E2F4_T | 0.910 | 0.50 |
| NAA25_T | 0.910 | 0.63 |
| NIP7_T | 0.910 | 0.77 |
| NPM3_T | 0.910 | 0.69 |
| PHB2_T | 0.910 | 0.72 |
| SNX21_T | 0.910 | −0.64 |
| UBE2I_T | 0.910 | 0.34 |
| CAPN15_T | 0.905 | 0.64 |
| HNRNPA1_T | 0.905 | 0.85 |
| SNHG4_T | 0.905 | 0.70 |
| SNORA52_T | 0.905 | 0.39 |
| SRSF12_Me | 0.905 | 1.50 |
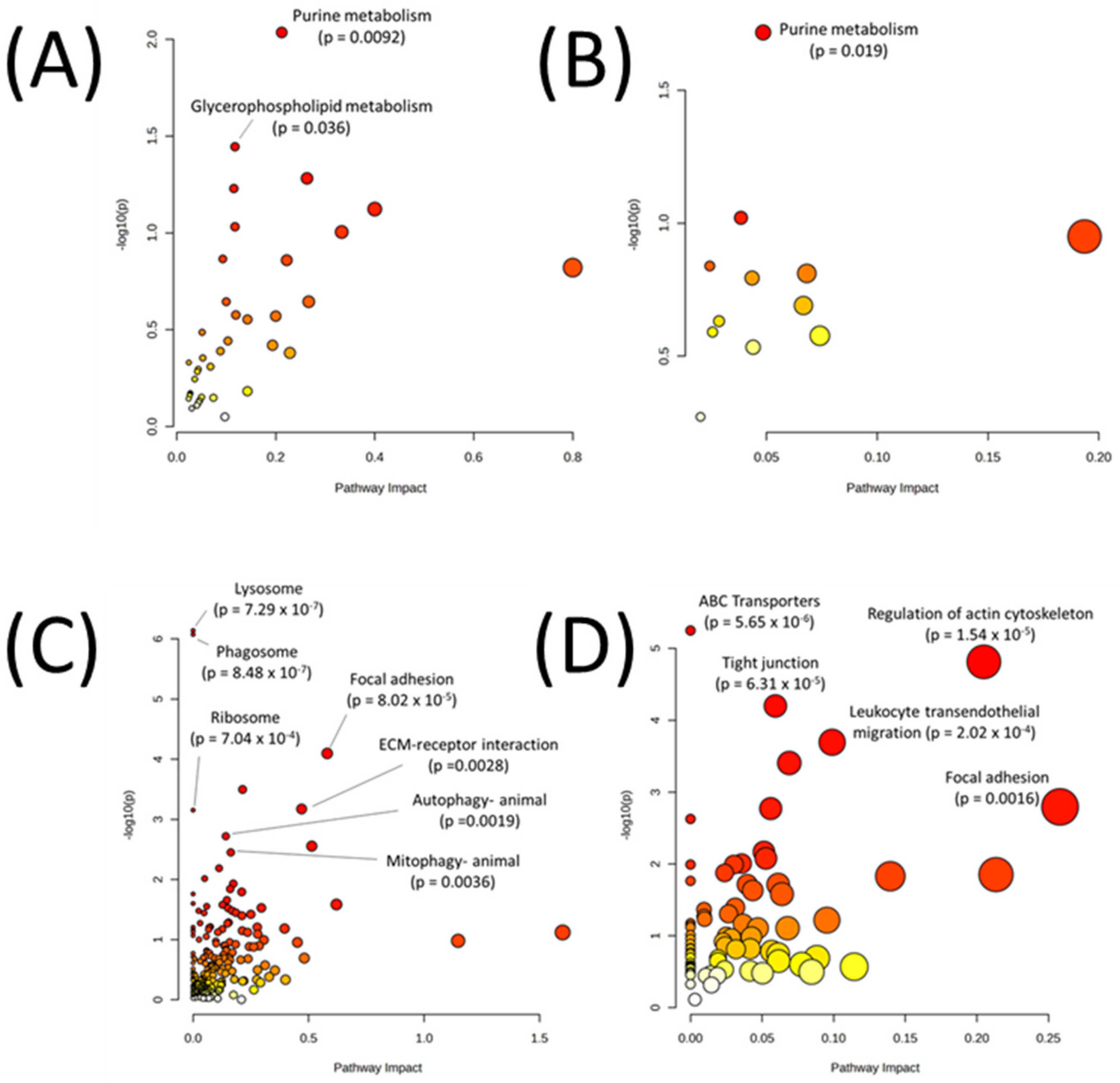
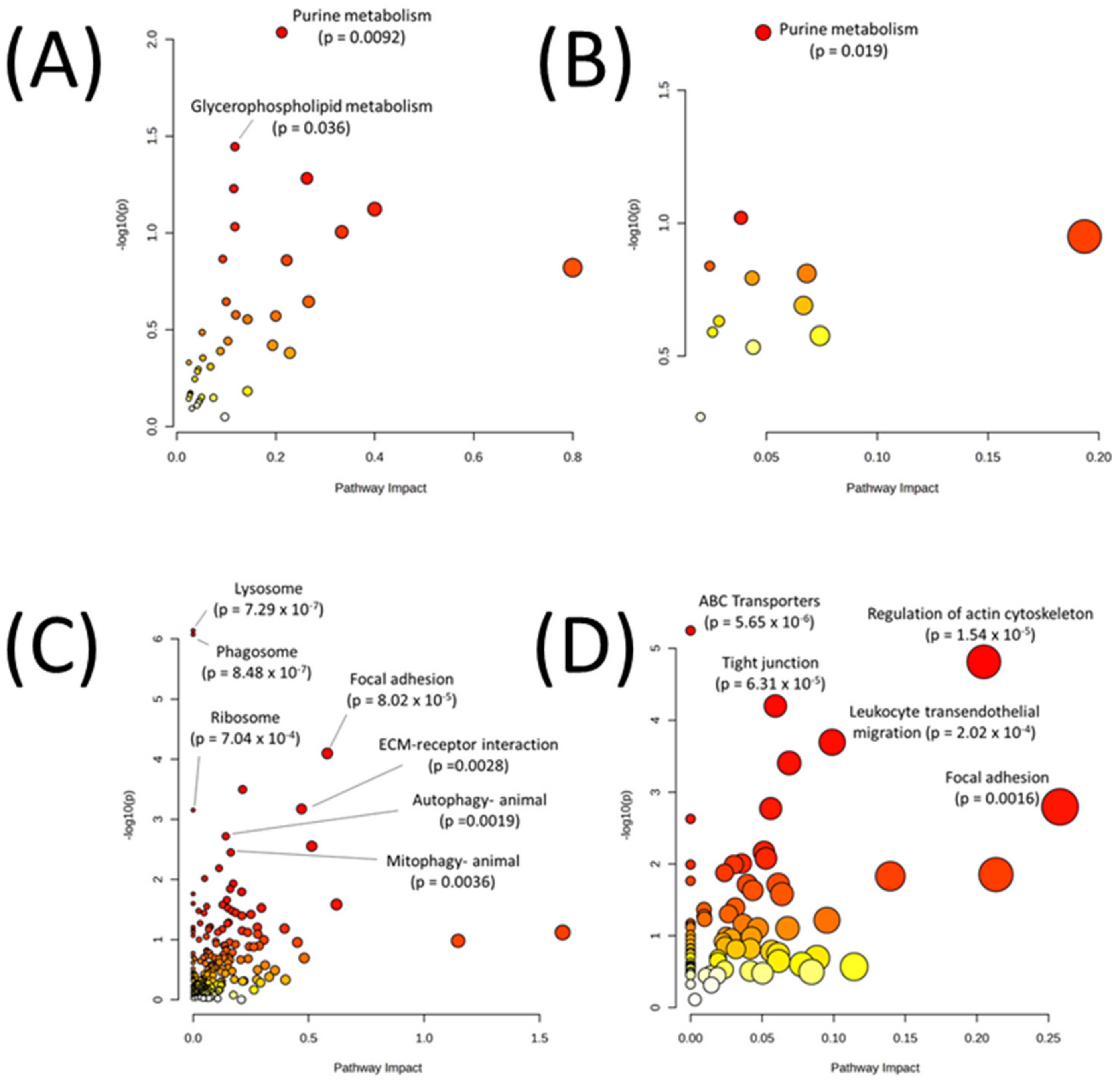
| Metabolic Pathways Only | |
|---|---|
| Pathway | p-Value |
| Purine metabolism | 0.009219 |
| Glycerophospholipid metabolism | 0.035887 |
| Sphingolipid metabolism | 0.052286 |
| Arginine biosynthesis | 0.058987 |
| One carbon pool by folate | 0.075348 |
| Glycerolipid metabolism | 0.092975 |
| Glycosaminoglycan biosynthesis—heparan sulfate/heparin | 0.098934 |
| Glycosaminoglycan degradation | 0.136300 |
| Nitrogen metabolism | 0.138360 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 0.151120 |
| All Pathways | |
| Pathway | p-value |
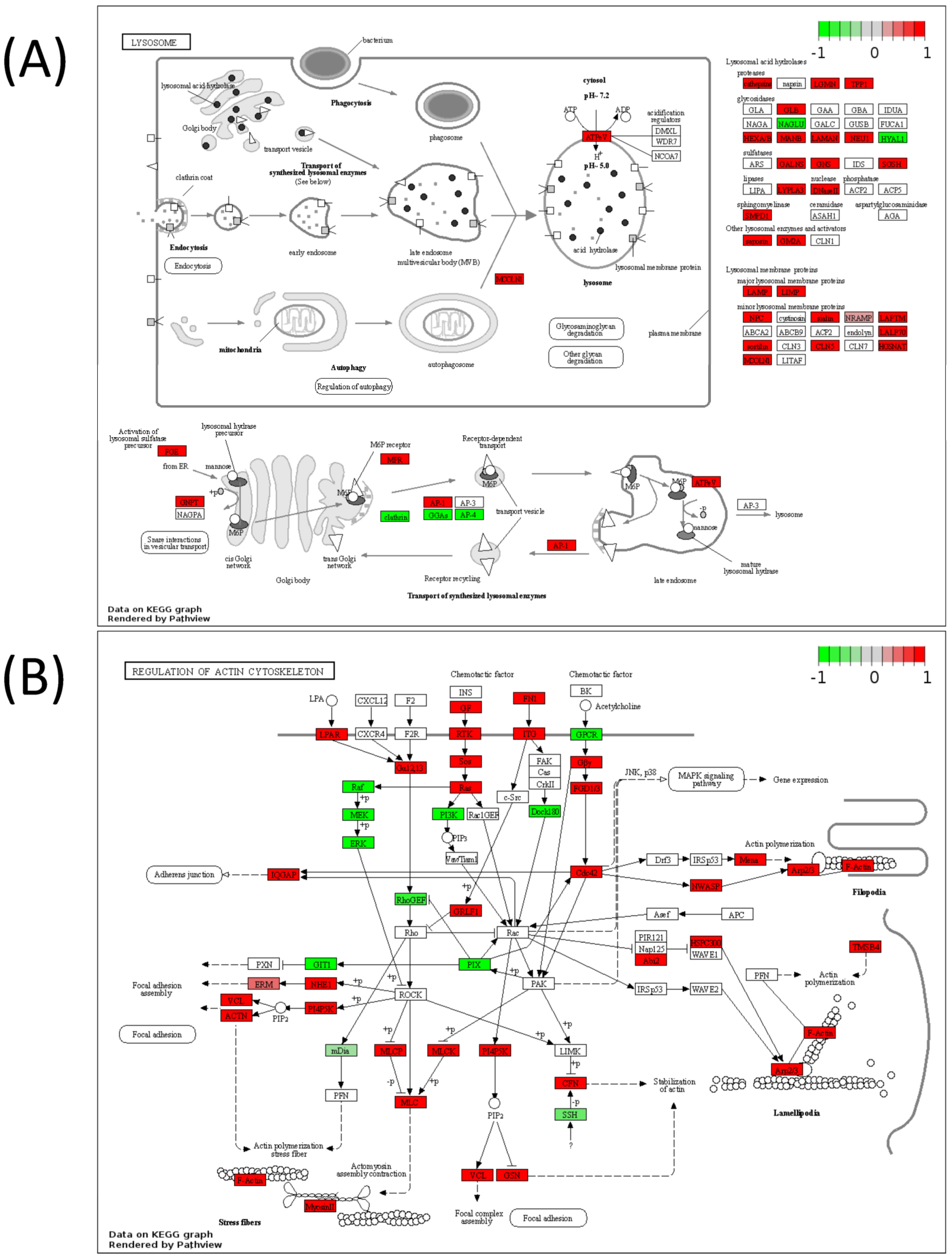
| Lysosome | 7.29 × 10−7 |
| Phagosome | 8.48 × 10−7 |
| Focal adhesion | 8.02 × 10−5 |
| Vibrio cholerae infection | 0.000319 |
| Regulation of actin cytoskeleton | 0.000673 |
| Ribosome | 0.000704 |
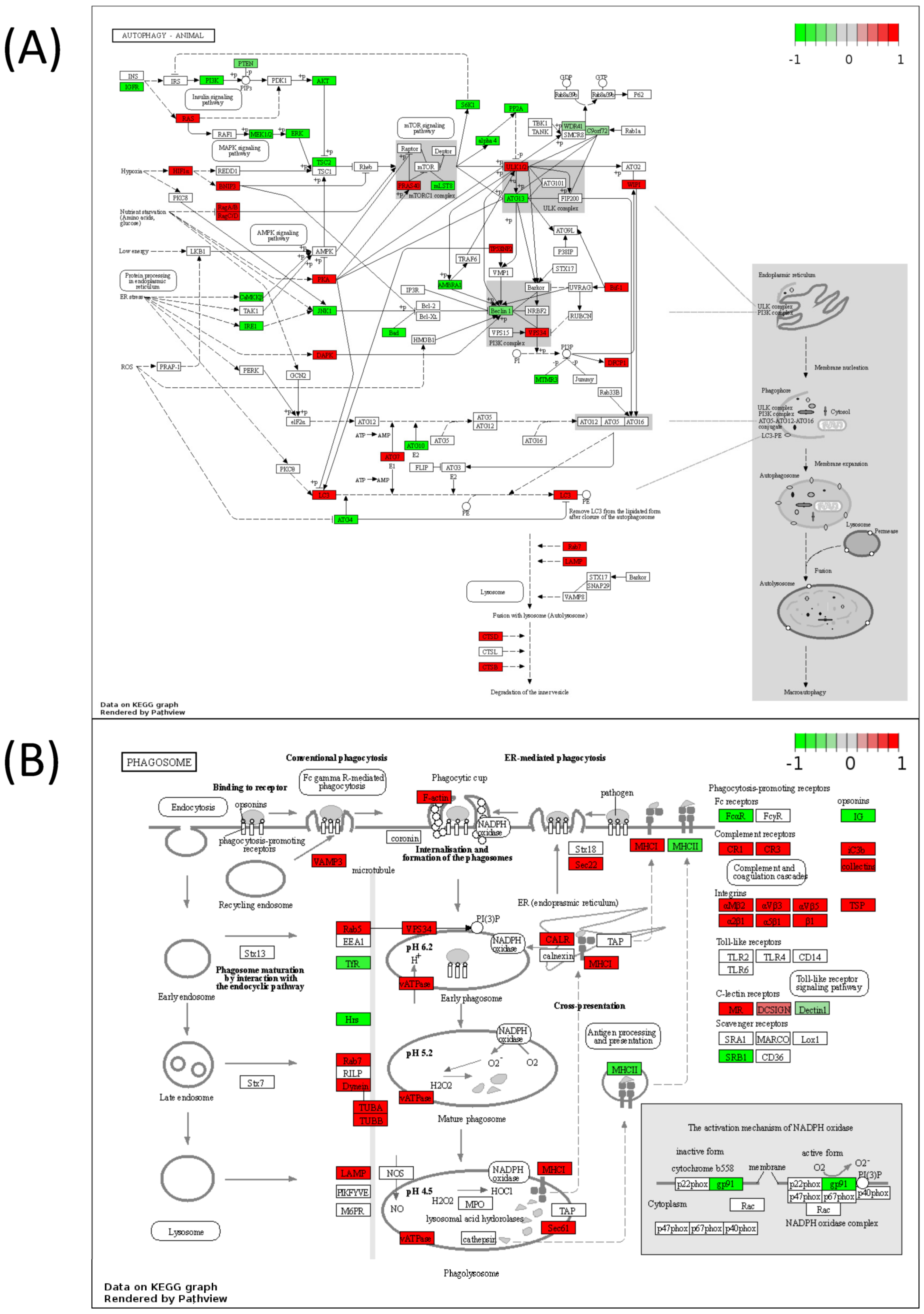
| Autophagy—animal | 0.00191 |
| ECM-receptor interaction | 0.00279 |
| Mitophagy—animal | 0.003552 |
| Human papillomavirus infection | 0.006515 |
| Metabolic Pathways Only | |
|---|---|
| Pathway | p-Value |
| Purine metabolism | 0.019147 |
| Arginine biosynthesis | 0.095444 |
| Pentose and glucuronate interconversions | 0.112200 |
| Citrate cycle (TCA cycle) | 0.144890 |
| Pyruvate metabolism | 0.154490 |
| Pentose phosphate pathway | 0.160830 |
| Glycolysis or Gluconeogenesis | 0.204070 |
| Cysteine and methionine metabolism | 0.233730 |
| Amino sugar and nucleotide sugar metabolism | 0.256740 |
| Steroid biosynthesis | 0.265210 |
| All Pathways | |
| Pathway | p-value |
| ABC transporters | 5.65 × 10−6 |
| Regulation of actin cytoskeleton | 1.54 × 10−5 |
| Tight junction | 6.31 × 10−5 |
| Leukocyte transendothelial migration | 0.000202 |
| Platelet activation | 0.000393 |
| Focal adhesion | 0.001603 |
| Pathogenic Escherichia coli infection | 0.001693 |
| Ribosome biogenesis in eukaryotes | 0.002361 |
| Phagosome | 0.006792 |
| Oxytocin signaling pathway | 0.008358 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rushing, B.R. Unlocking the Molecular Secrets of Antifolate Drug Resistance: A Multi-Omics Investigation of the NCI-60 Cell Line Panel. Biomedicines 2023, 11, 2532. https://doi.org/10.3390/biomedicines11092532
Rushing BR. Unlocking the Molecular Secrets of Antifolate Drug Resistance: A Multi-Omics Investigation of the NCI-60 Cell Line Panel. Biomedicines. 2023; 11(9):2532. https://doi.org/10.3390/biomedicines11092532
Chicago/Turabian StyleRushing, Blake R. 2023. "Unlocking the Molecular Secrets of Antifolate Drug Resistance: A Multi-Omics Investigation of the NCI-60 Cell Line Panel" Biomedicines 11, no. 9: 2532. https://doi.org/10.3390/biomedicines11092532
APA StyleRushing, B. R. (2023). Unlocking the Molecular Secrets of Antifolate Drug Resistance: A Multi-Omics Investigation of the NCI-60 Cell Line Panel. Biomedicines, 11(9), 2532. https://doi.org/10.3390/biomedicines11092532