

The Multi-Kinase Inhibitor RepSox Enforces Barrier Function in the Face of Both VEGF and Cytokines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Electrical Cell-Substrate Impedance Sensing (ECIS)
2.4. Gelatin Trapping Assay (GTA)
2.5. Small Interfering RNA (siRNA)
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
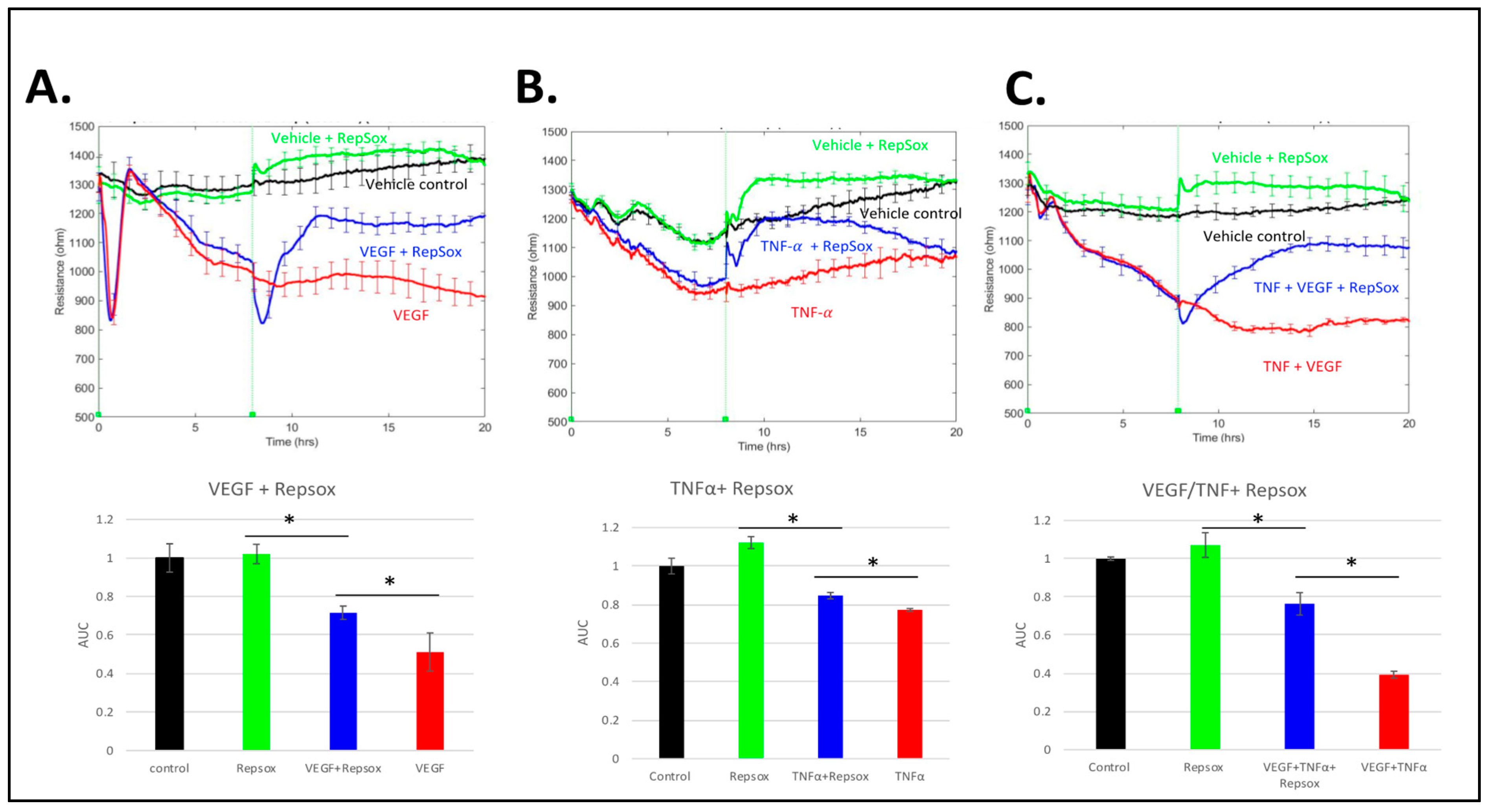
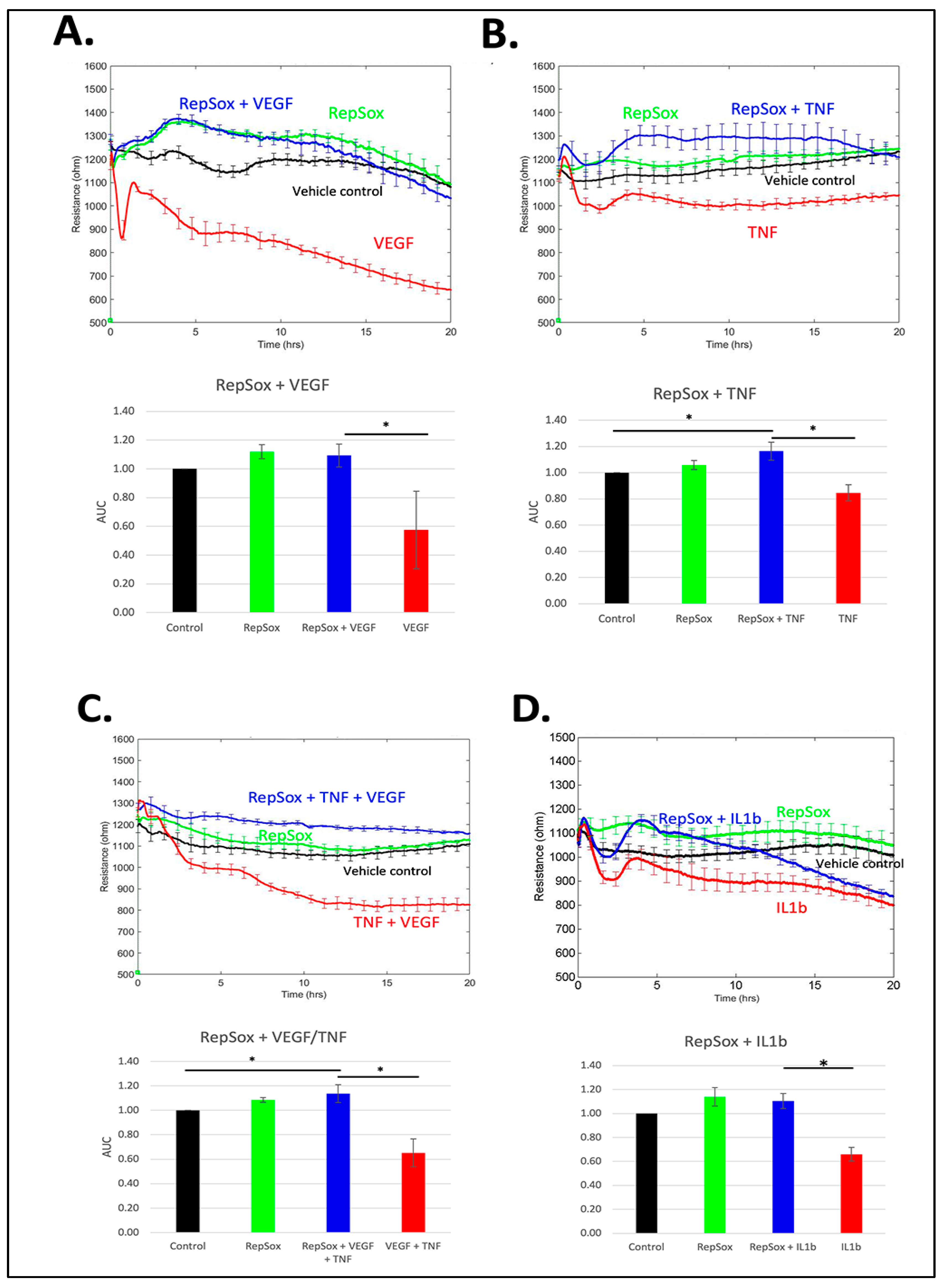
3.1. RS Prevented Barrier Relaxation by Distinct Agonists
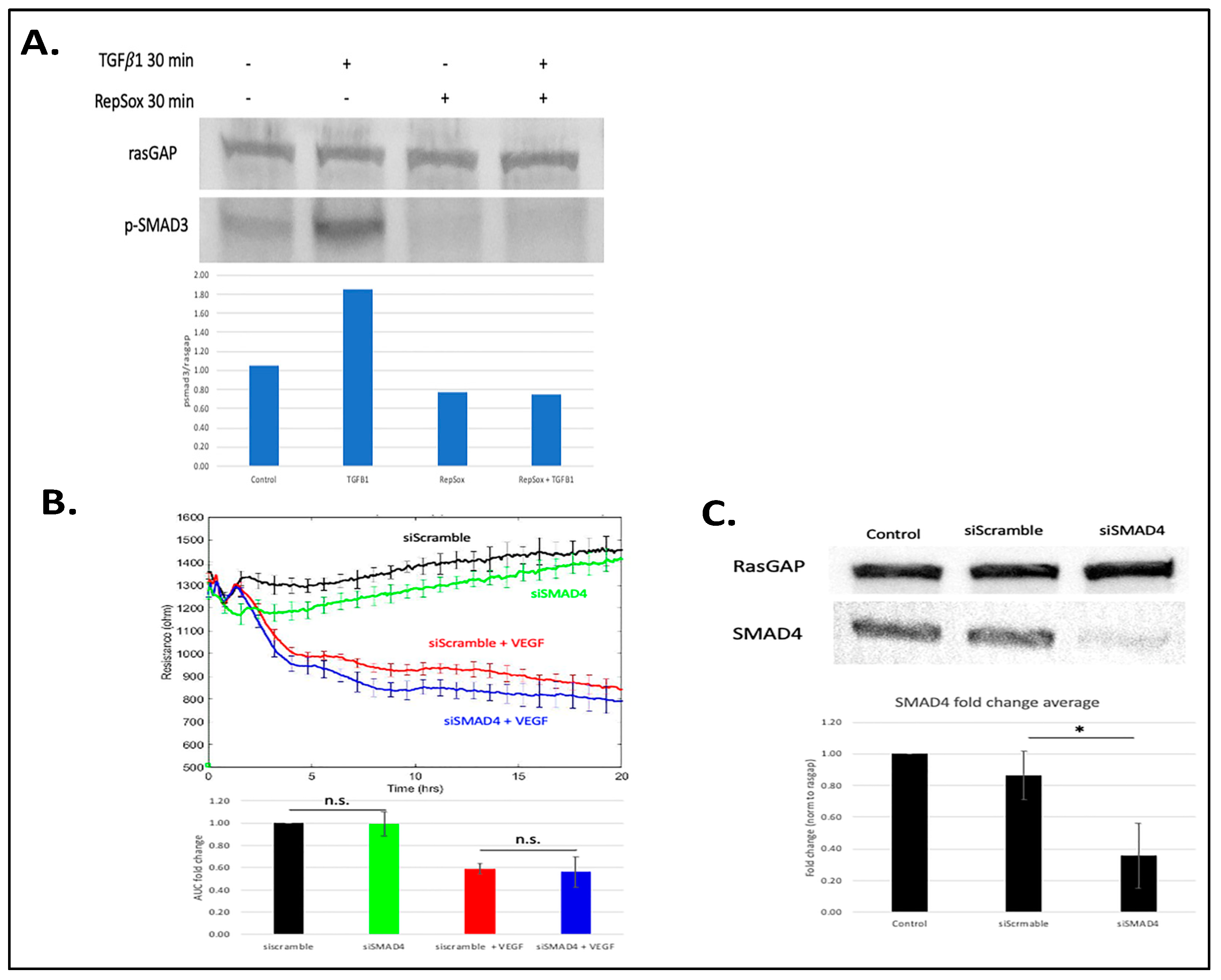
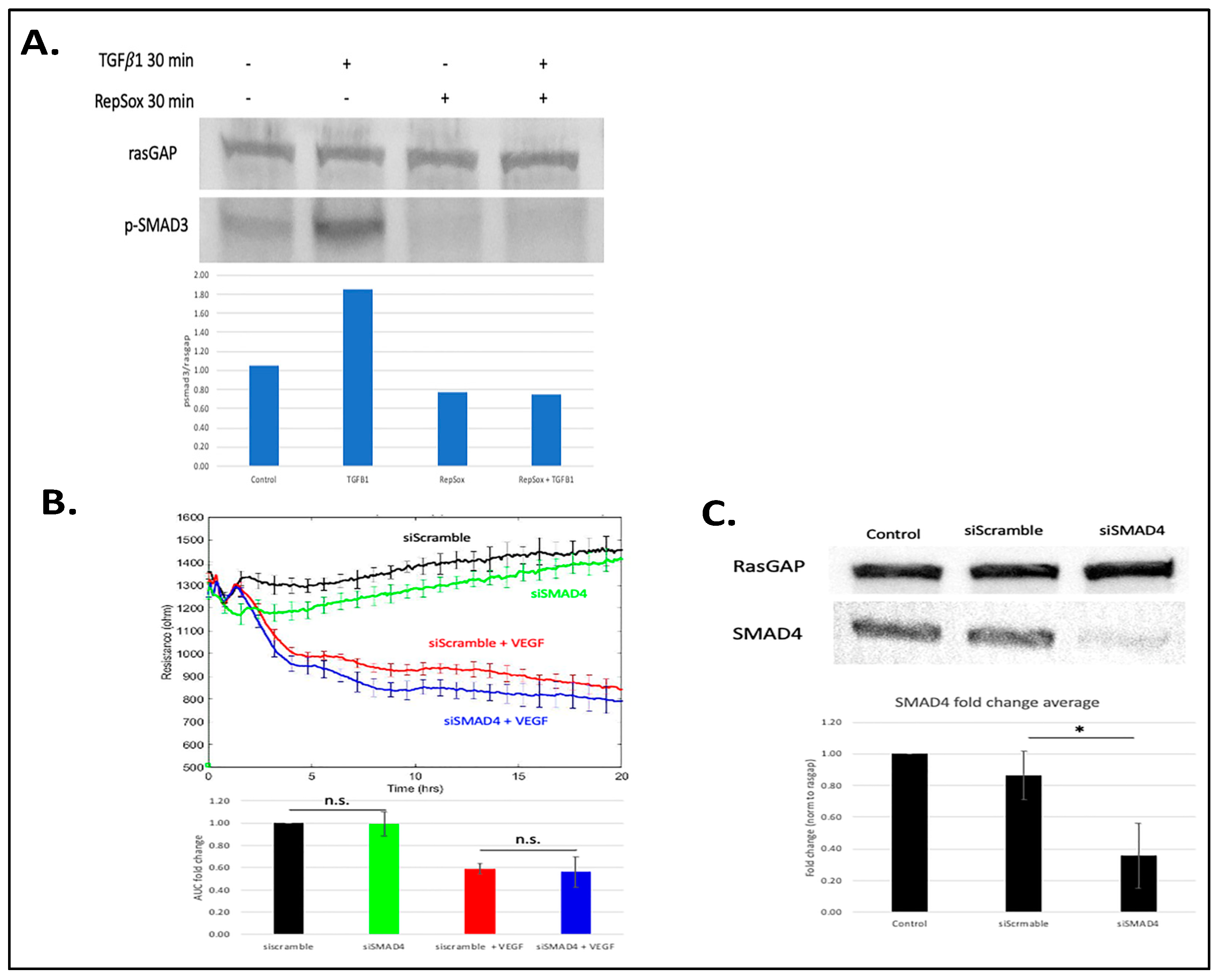
3.2. Antagonizing TGFβ Signaling Was Insufficient for the RS Effect
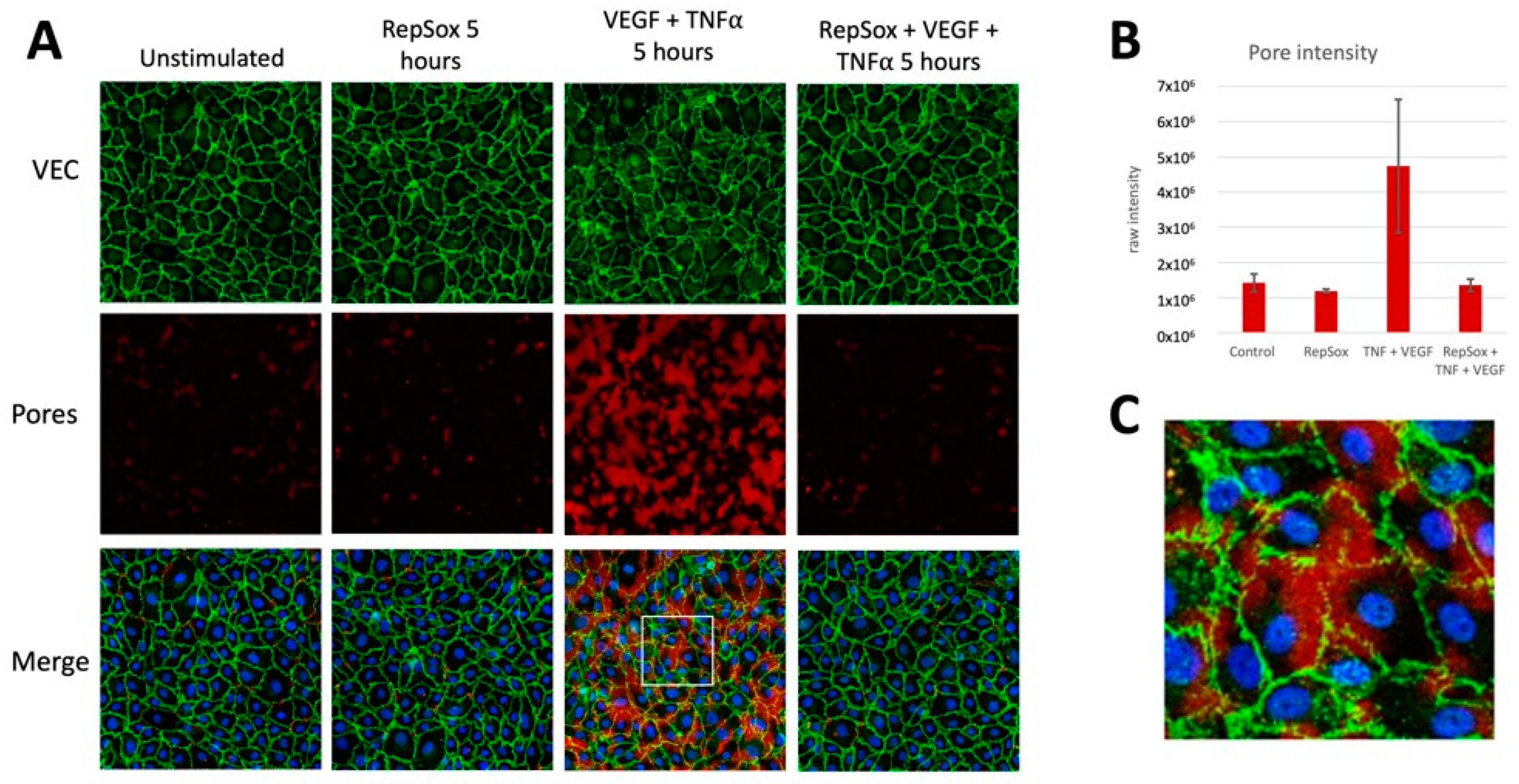
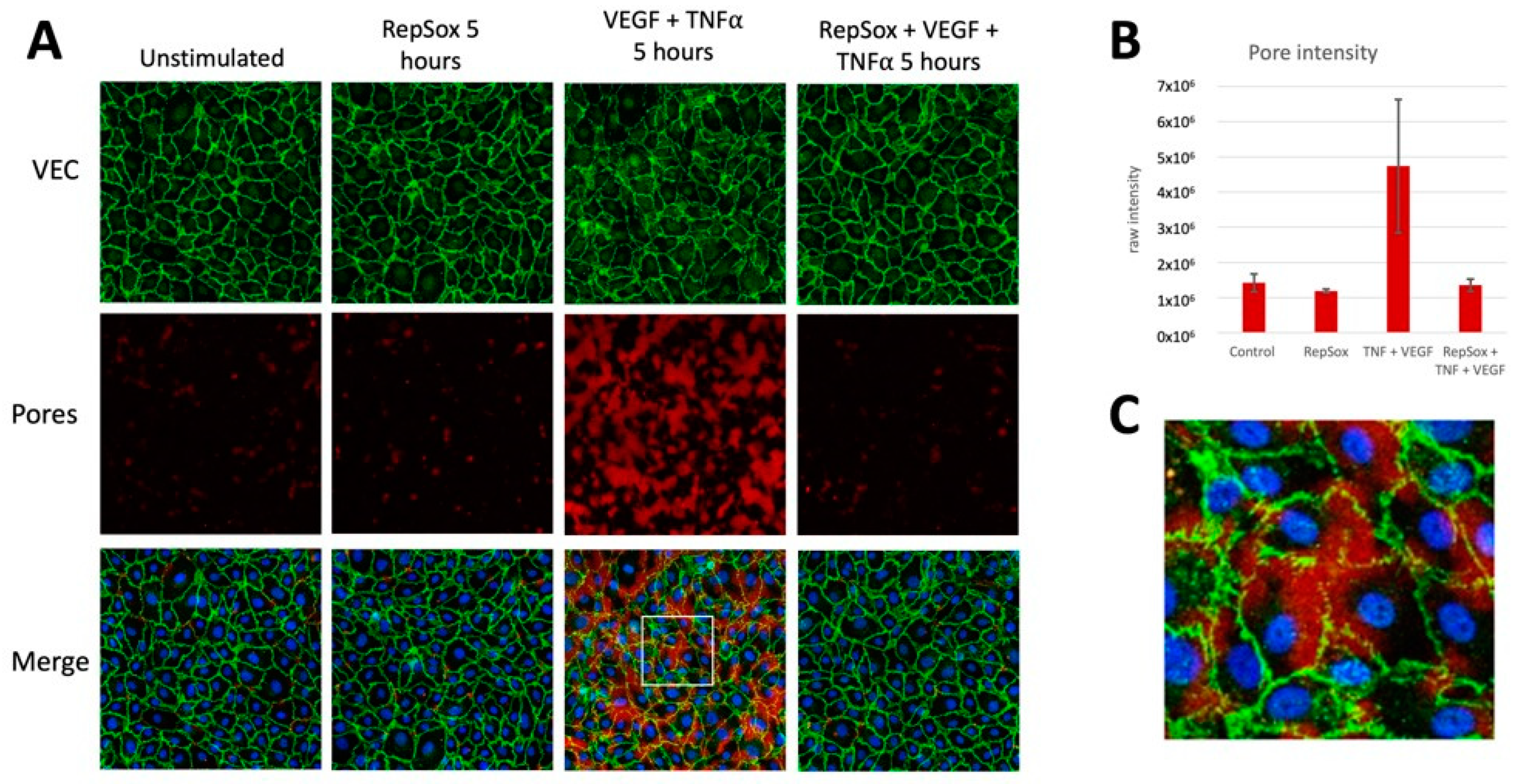
3.3. RS Prevented Agonist-Induced Pore Formation and VE-Cadherin Disorganization
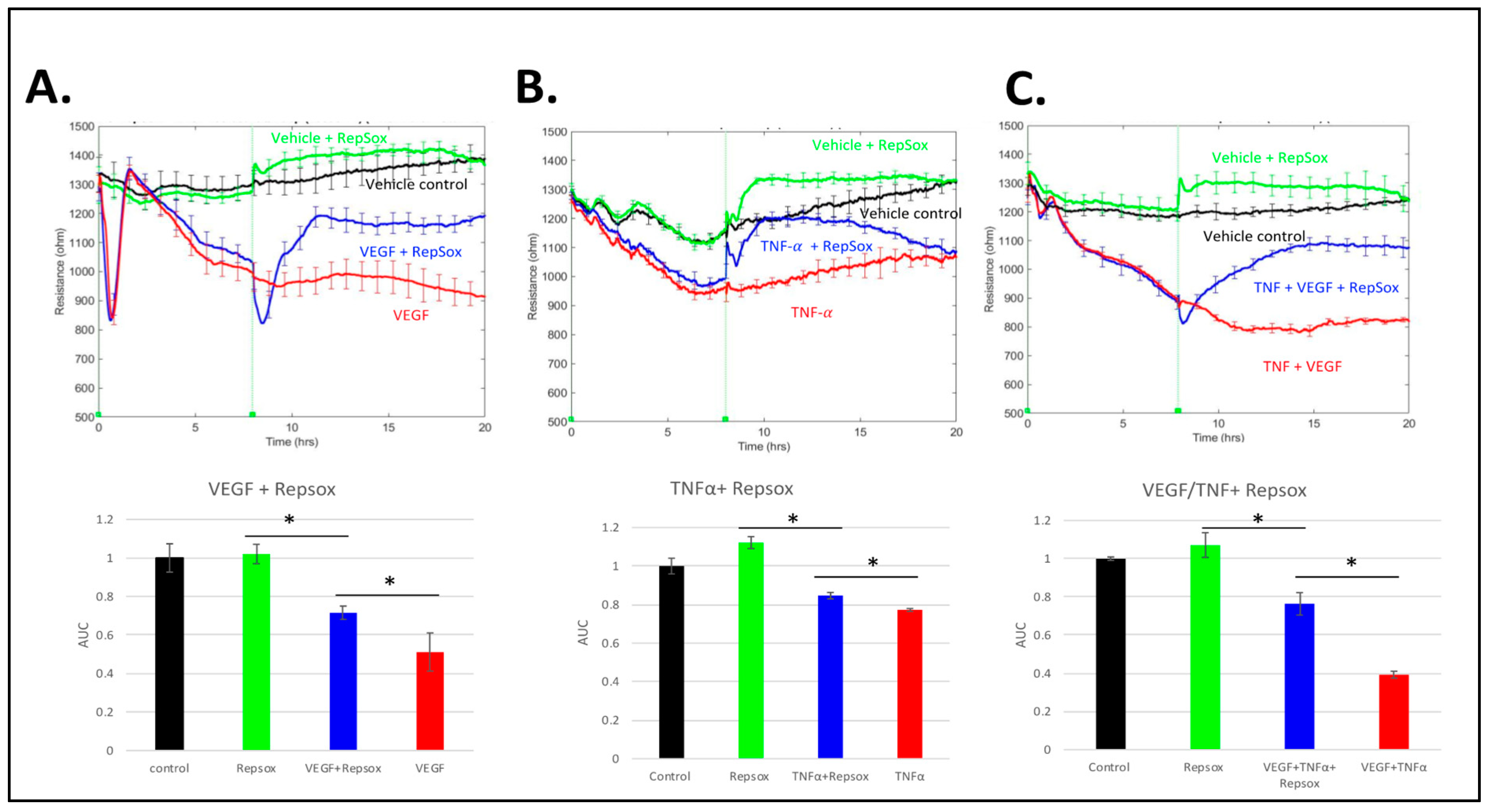
3.4. RS Reclosed the Barrier Relaxed by Distinct Agonists
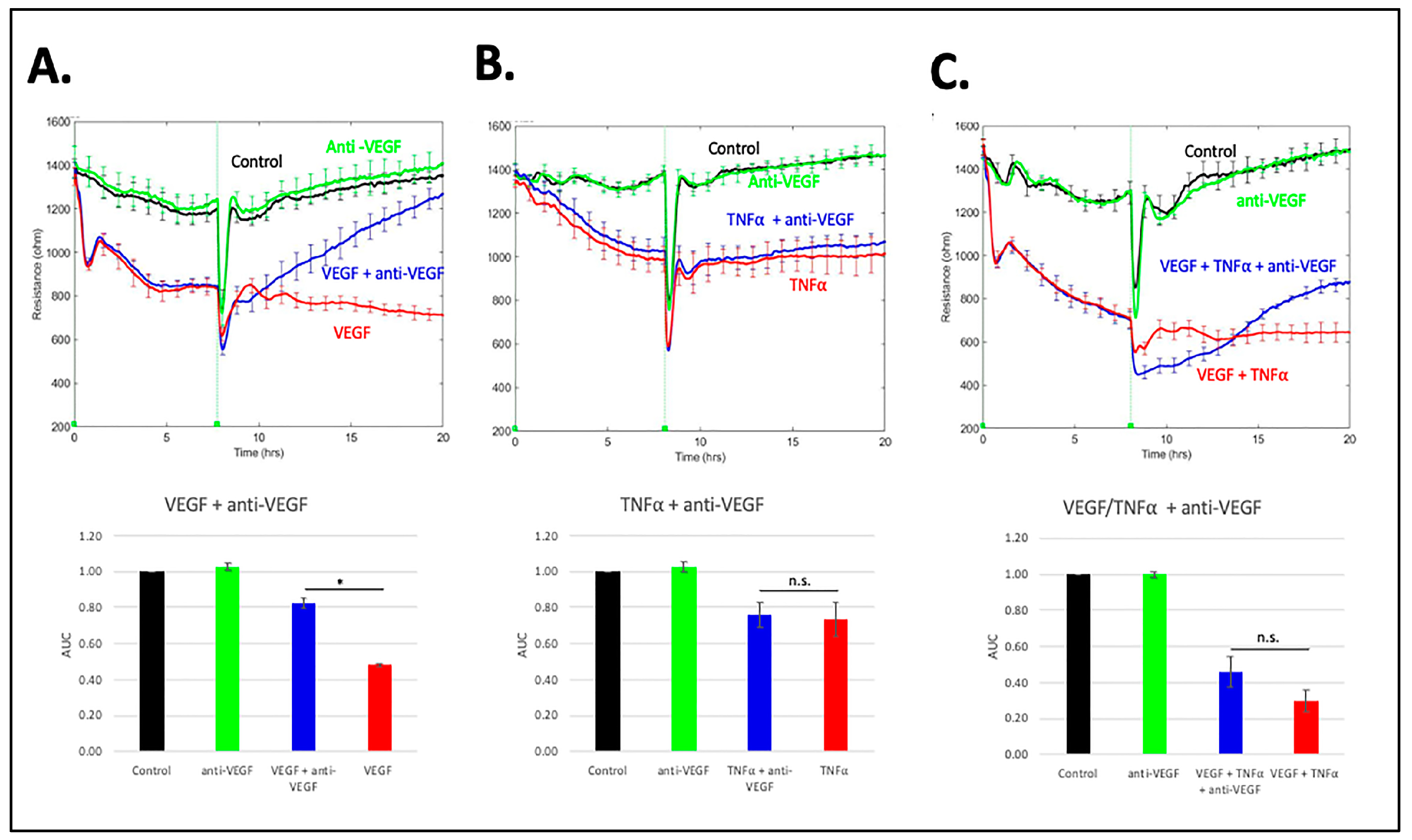
3.5. Comparison of RS with Anti-VEGF
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuo, C.Y.J.; Murphy, R.; Rupenthal, I.D.; Mugisho, O.O. Correlation between the progression of diabetic retinopathy and inflammasome biomarkers in vitreous and serum—A systematic review. BMC Ophthalmol. 2022, 22, 238. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef]
- Ellis, M.P.; Lent-Schochet, D.; Lo, T.; Yiu, G. Emerging Concepts in the Treatment of Diabetic Retinopathy. Curr. Diabetes Rep. 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Roudnicky, F.; Zhang, J.D.; Kim, B.K.; Pandya, N.J.; Lan, Y.; Sach-Peltason, L.; Ragelle, H.; Strassburger, P.; Gruener, S.; Lazendic, M.; et al. Inducers of the endothelial cell barrier identified through chemogenomic screening in genome-edited hPSC-endothelial cells. Proc. Natl. Acad. Sci. USA 2020, 117, 19854–19865. [Google Scholar] [CrossRef]
- Gellibert, F.; Woolven, J.; Fouchet, M.H.; Mathews, N.; Goodland, H.; Lovegrove, V.; Laroze, A.; Nguyen, V.L.; Sautet, S.; Wang, R.; et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J. Med. Chem. 2004, 47, 4494–4506. [Google Scholar] [CrossRef]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef]
- Valius, M.; Bazenet, C.; Kazlauskas, A. Tyrosines 1021 and 1009 are phosphorylation sites in the carboxy terminus of the platelet-derived growth factor receptor b subunit and are required for binding of phospholipase Cg and a 64-kilodalton protein, respectively. Mol. Cell. Biol. 1993, 13, 133–143. [Google Scholar]
- Li, Y.; Yan, Z.; Chaudhry, K.; Kazlauskas, A. The Renin-Angiotensin-Aldosterone System (RAAS) Is One of the Effectors by Which Vascular Endothelial Growth Factor (VEGF)/Anti-VEGF Controls the Endothelial Cell Barrier. Am. J. Pathol. 2020, 190, 1971–1981. [Google Scholar] [CrossRef]
- Dubrovskyi, O.; Birukova, A.A.; Birukov, K.G. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Investig. 2013, 93, 254–263. [Google Scholar] [CrossRef]
- Serikbaeva, A.; Li, Y.; Ganesh, B.; Zelkha, R.; Kazlauskas, A. Hyperglycemia Promotes Mitophagy and Thereby Mitigates Hyperglycemia-Induced Damage. Am. J. Pathol. 2022, 192, 1779–1794. [Google Scholar] [CrossRef]
- He, D.; Gao, J.; Zheng, L.; Liu, S.; Ye, L.; Lai, H.; Pan, B.; Pan, W.; Lou, C.; Chen, Z.; et al. TGF-beta inhibitor RepSox suppresses osteosarcoma via the JNK/Smad3 signaling pathway. Int. J. Oncol. 2021, 59, 84. [Google Scholar] [CrossRef] [PubMed]
- Baccouche, B.; Lietuvninkas, L.; Kazlauskas, A. Activin A Limits VEGF-Induced Permeability via VE-PTP. Int. J. Mol. Sci. 2023, 24, 8698. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhu, H.; Li, X.; Ma, C.; Li, Y.; Sun, T.; Wang, Y.; Wang, C.; Guan, W.; Liu, C. RepSox effectively promotes the induced differentiation of sheep fibroblasts into adipocytes via the inhibition of the TGF-beta1/Smad pathway. Int. J. Mol. Med. 2021, 48, 148. [Google Scholar] [CrossRef]
- Shi, C.J.; Lian, J.J.; Zhang, B.W.; Cha, J.X.; Hua, Q.H.; Pi, X.P.; Hou, Y.J.; Xie, X.; Zhang, R. TGFbetaR-1/ALK5 inhibitor RepSox induces enteric glia-to-neuron transition and influences gastrointestinal mobility in adult mice. Acta Pharmacol. Sin. 2023, 44, 92–104. [Google Scholar] [CrossRef]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Simons, M. An inside view: VEGF receptor trafficking and signaling. Physiology 2012, 27, 213–222. [Google Scholar] [CrossRef]
- Schneider-Brachert, W.; Tchikov, V.; Neumeyer, J.; Jakob, M.; Winoto-Morbach, S.; Held-Feindt, J.; Heinrich, M.; Merkel, O.; Ehrenschwender, M.; Adam, D.; et al. Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity 2004, 21, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Reschke, C.R.; Reddy, A.; Mae, M.A.; Connolly, R.; Behan, C.; O’Keeffe, E.; Bolger, I.; Hudson, N.; et al. Microvascular stabilization via blood-brain barrier regulation prevents seizure activity. Nat. Commun. 2022, 13, 2003. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lietuvninkas, L.; Baccouche, B.; Kazlauskas, A. The Multi-Kinase Inhibitor RepSox Enforces Barrier Function in the Face of Both VEGF and Cytokines. Biomedicines 2023, 11, 2431. https://doi.org/10.3390/biomedicines11092431
Lietuvninkas L, Baccouche B, Kazlauskas A. The Multi-Kinase Inhibitor RepSox Enforces Barrier Function in the Face of Both VEGF and Cytokines. Biomedicines. 2023; 11(9):2431. https://doi.org/10.3390/biomedicines11092431
Chicago/Turabian StyleLietuvninkas, Lina, Basma Baccouche, and Andrius Kazlauskas. 2023. "The Multi-Kinase Inhibitor RepSox Enforces Barrier Function in the Face of Both VEGF and Cytokines" Biomedicines 11, no. 9: 2431. https://doi.org/10.3390/biomedicines11092431
APA StyleLietuvninkas, L., Baccouche, B., & Kazlauskas, A. (2023). The Multi-Kinase Inhibitor RepSox Enforces Barrier Function in the Face of Both VEGF and Cytokines. Biomedicines, 11(9), 2431. https://doi.org/10.3390/biomedicines11092431