
The Expression of Epac2 and GluA3 in an Alzheimer’s Disease Experimental Model and Postmortem Patient Samples
 , , and
, , and 
Abstract
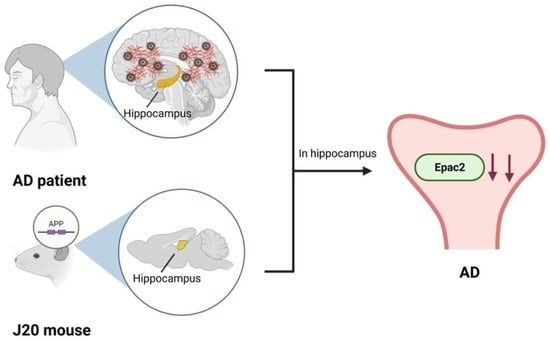
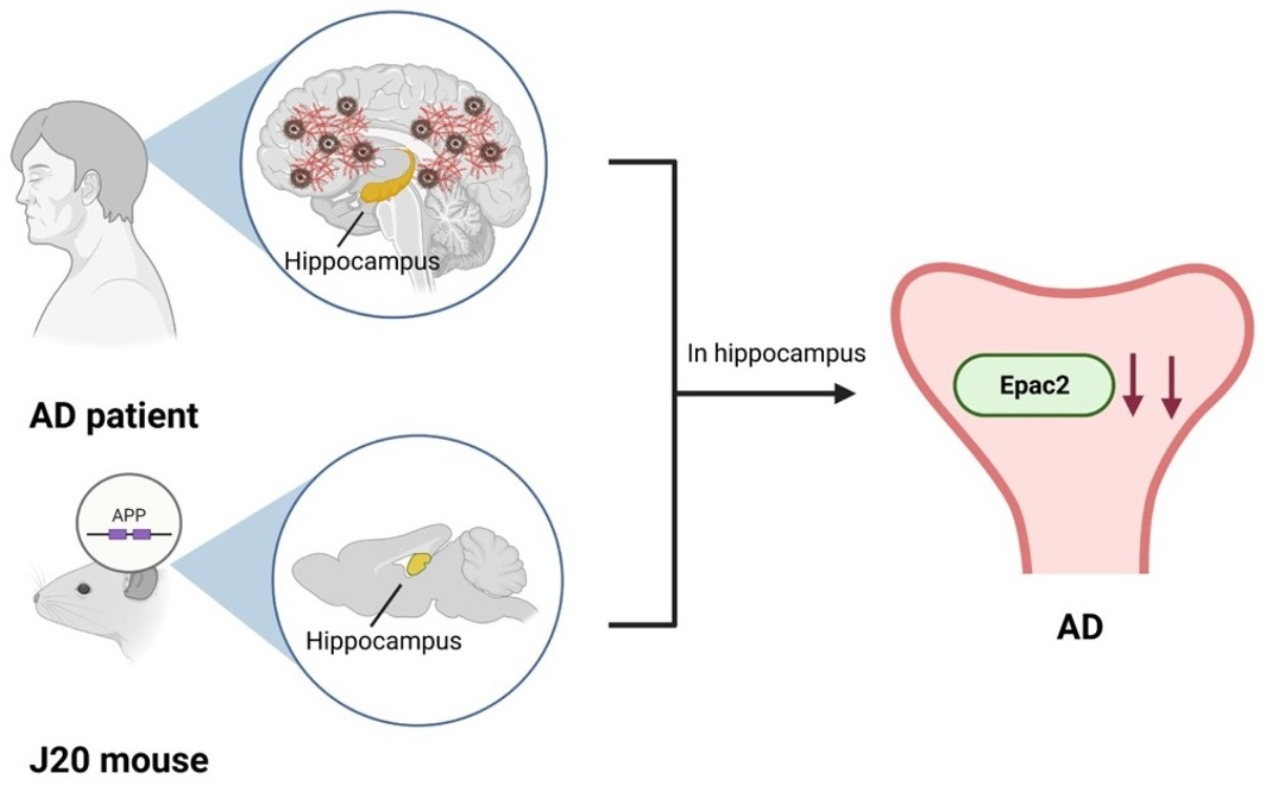{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. J20 Samples
2.3. Western Blot
2.4. Statistical Analysis
3. Results
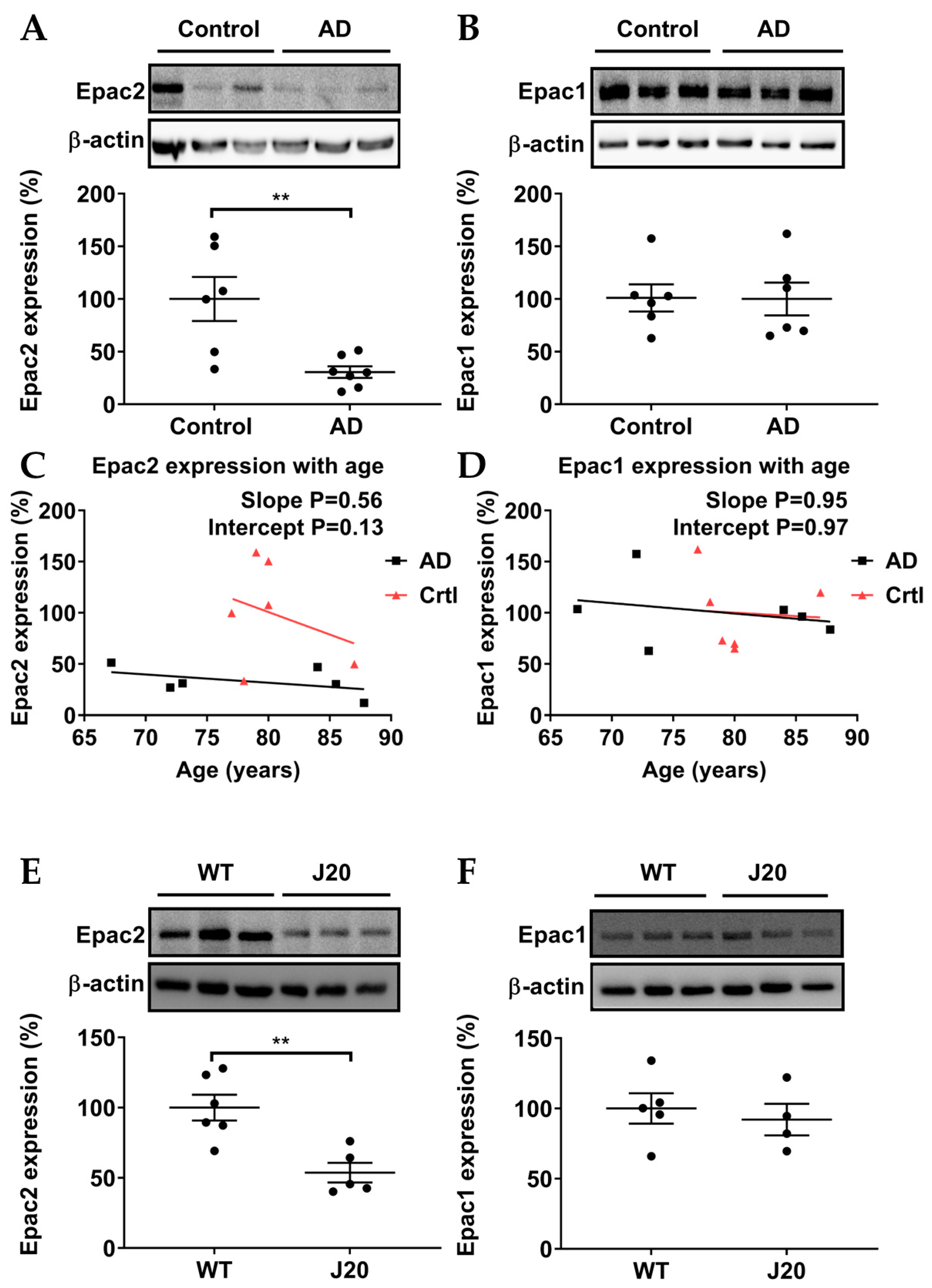
3.1. Epac2, but Not Epac1, Was Downregulated in AD Patients and AD Model Mice
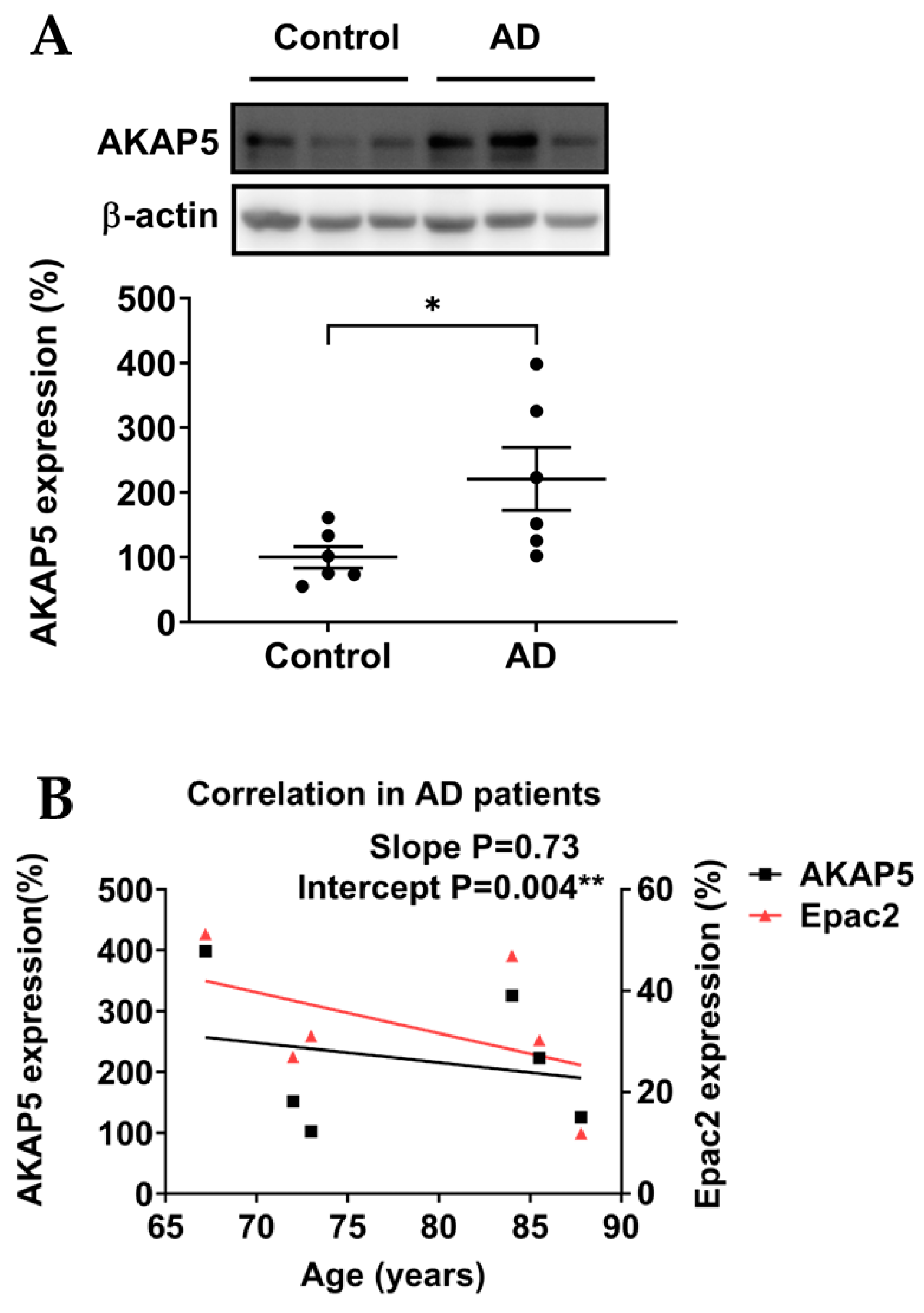
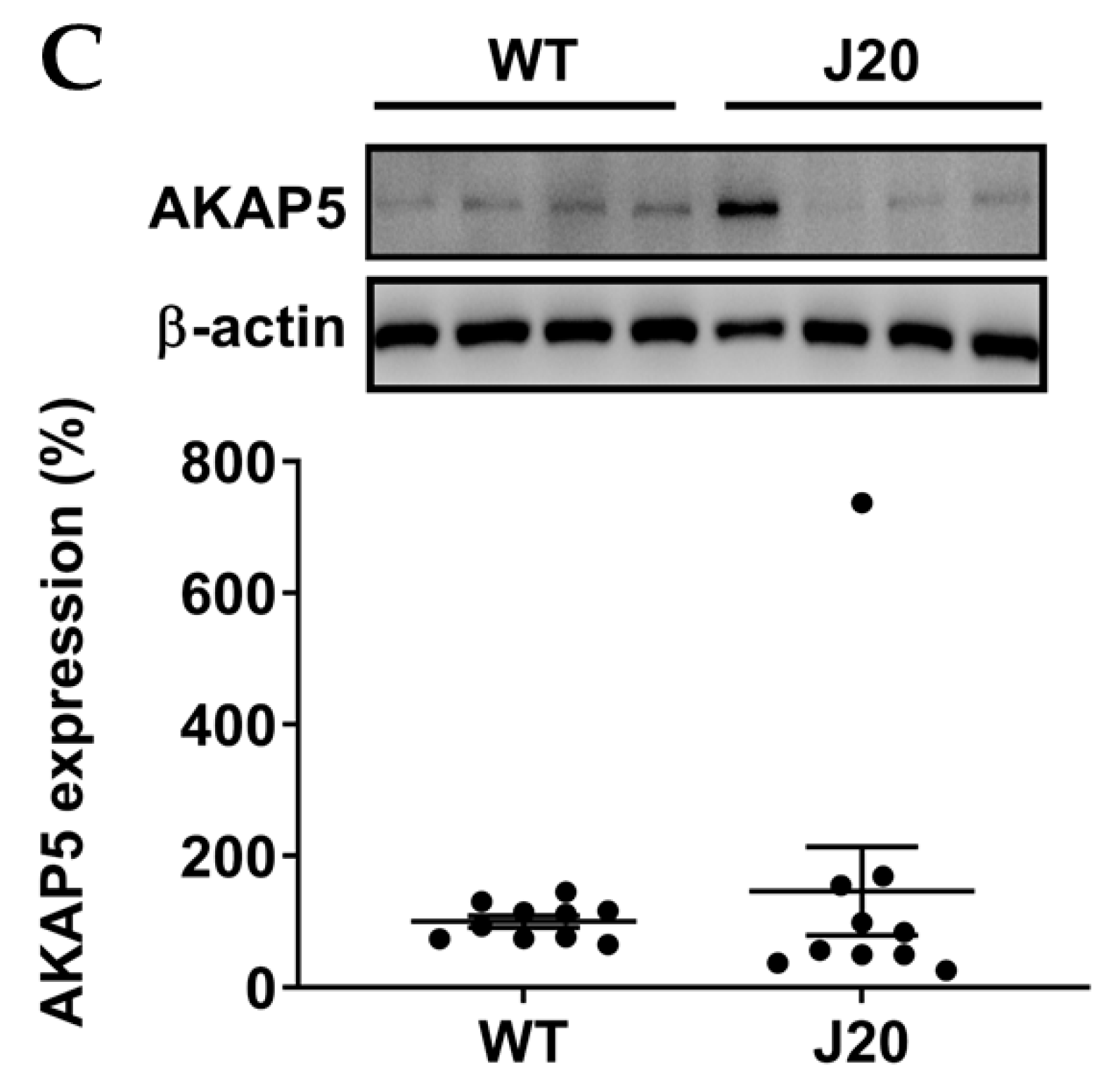
3.2. AKAP5 Was Upregulated in Postmortem Samples of AD Patients
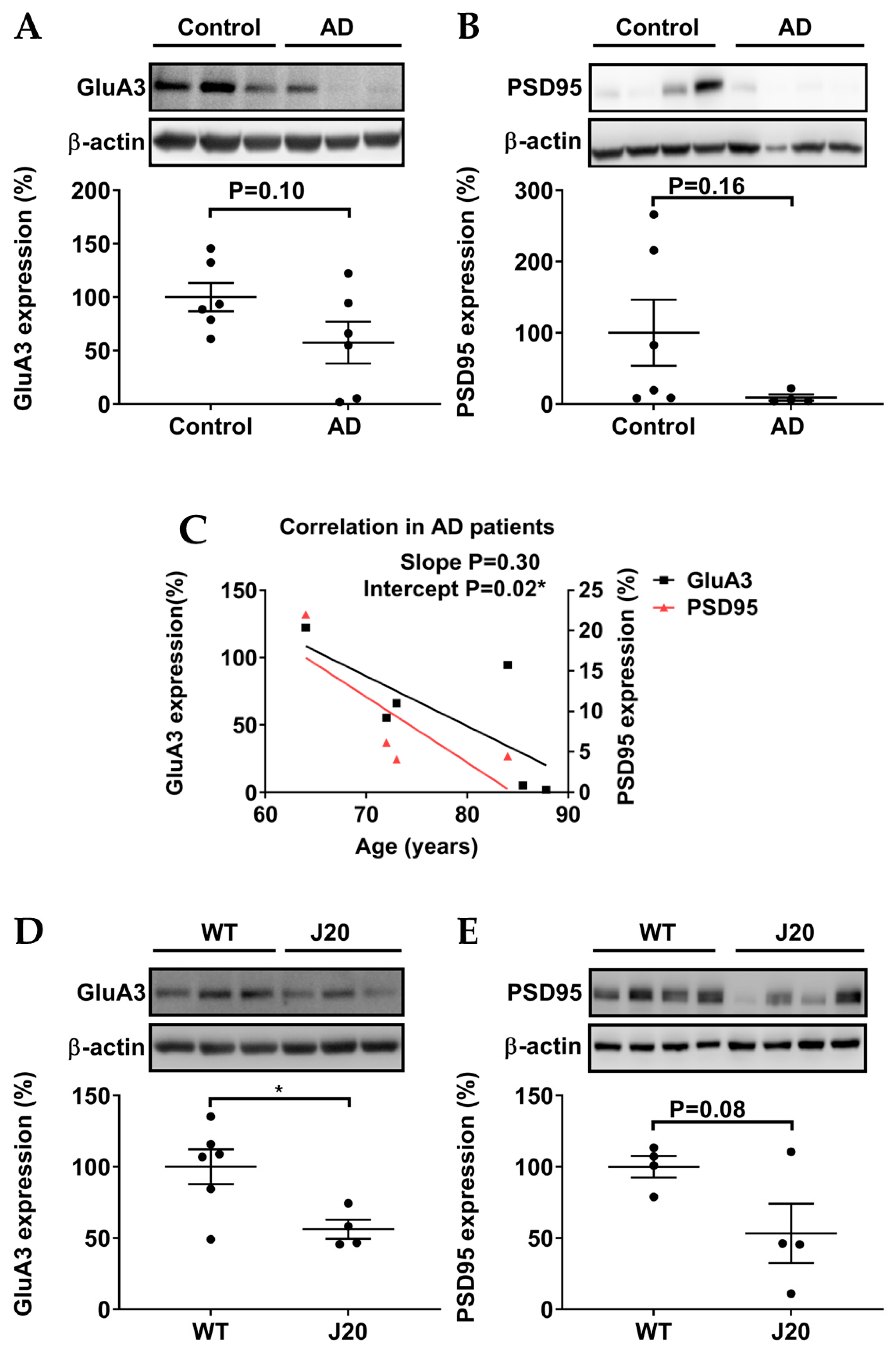
3.3. Protein Levels of GluA3 and PSD95 in AD Patients and AD Models
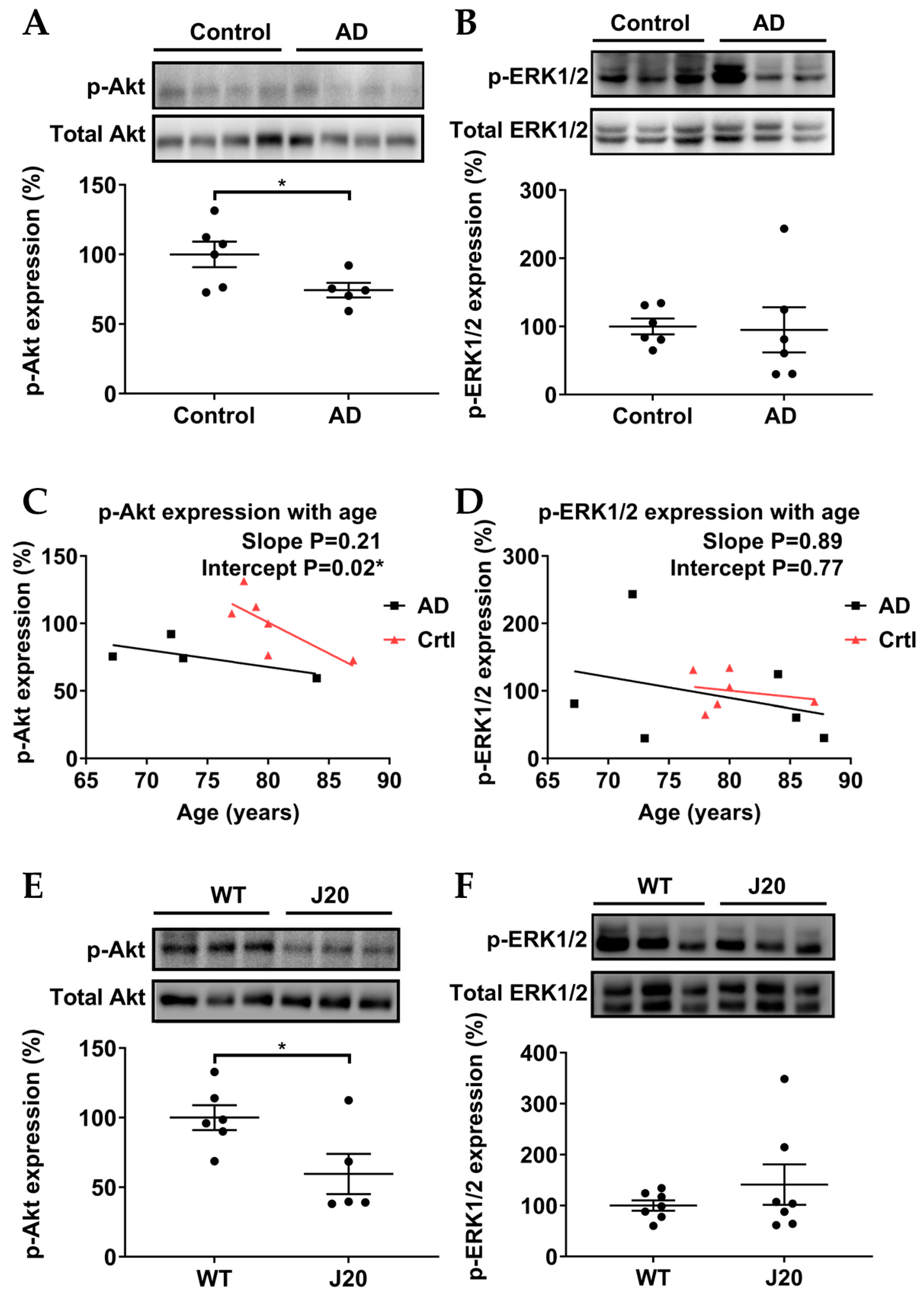
3.4. p-Akt (Ser473), but Not p-ERK1/2 (Thr202/Tyr204), Were Downregulated in AD Models
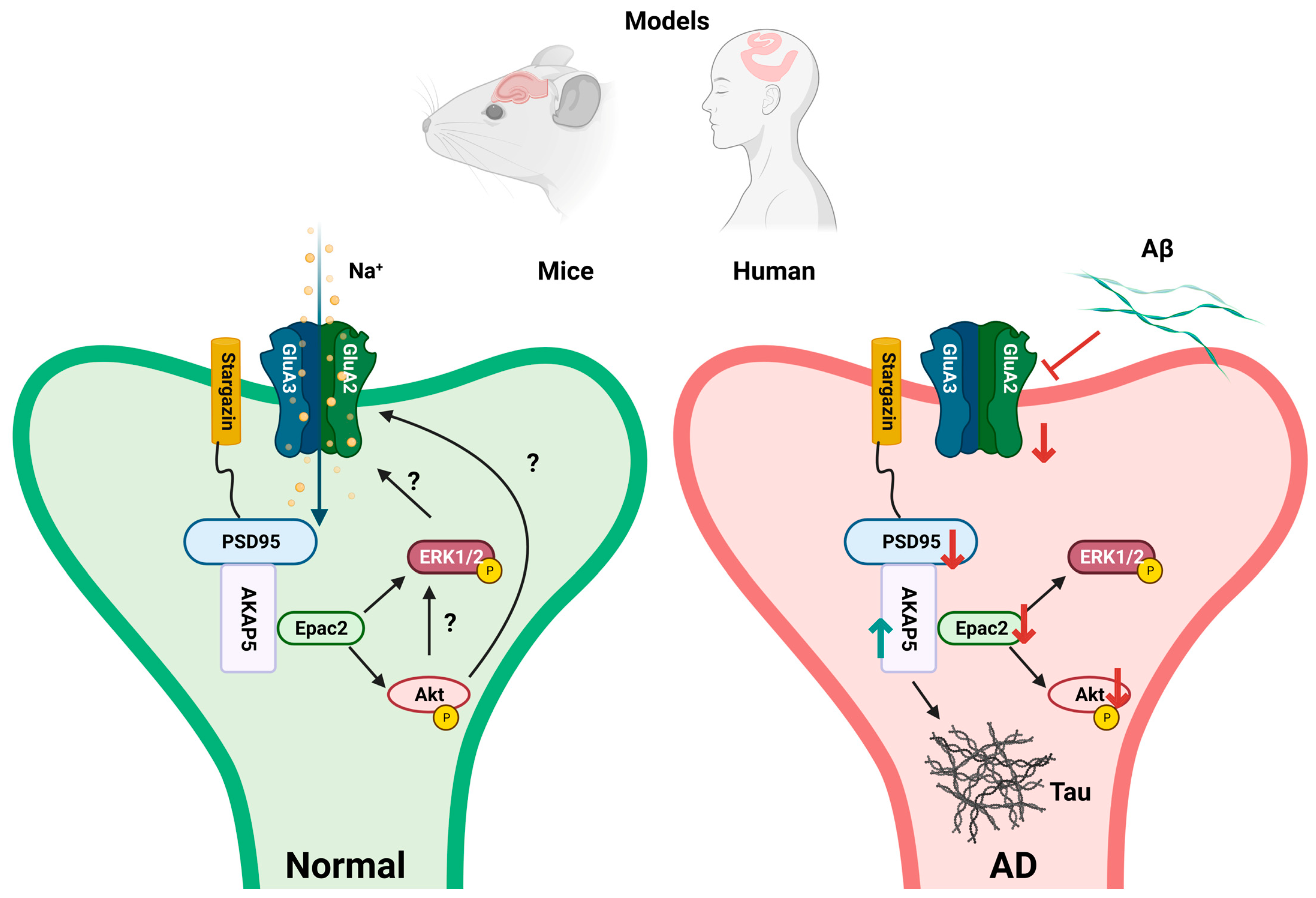
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Schmachtenberg, T.; Monsees, J.; Hoffmann, W.; van den Berg, N.; Stentzel, U.; Thyrian, J.R. Comparing national dementia plans and strategies in Europe—Is there a focus of care for people with dementia from a migration background? BMC Public Health 2020, 20, 784. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Rai, S.P.; Krohn, M.; Pahnke, J. Early Cognitive Training Rescues Remote Spatial Memory but Reduces Cognitive Flexibility in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2020, 75, 1301–1317. [Google Scholar] [CrossRef]
- Roy, D.S.; Arons, A.; Mitchell, T.I.; Pignatelli, M.; Ryan, T.J.; Tonegawa, S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 2016, 531, 508–512. [Google Scholar] [CrossRef]
- Bostancıklıoğlu, M. An update on memory formation and retrieval: An engram-centric approach. Alzheimer’s Dement. 2020, 16, 926–937. [Google Scholar] [CrossRef]
- Fernandes, H.B.; Riordan, S.; Nomura, T.; Remmers, C.L.; Kraniotis, S.; Marshall, J.J.; Kukreja, L.; Vassar, R.; Contractor, A. Epac2 Mediates cAMP-Dependent Potentiation of Neurotransmission in the Hippocampus. J. Neurosci. 2015, 35, 6544–6553. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Castellanos, N.; Da Silva-Matos, C.M.; Zhou, K.; Canto, C.B.; Renner, M.C.; Koene, L.M.; Ozyildirim, O.; Sprengel, R.; Kessels, H.W.; De Zeeuw, C.I. Motor Learning Requires Purkinje Cell Synaptic Potentiation through Activation of AMPA-Receptor Subunit GluA3. Neuron 2017, 93, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Ostroveanu, A.; van der Zee, E.A.; Eisel, U.L.; Schmidt, M.; Nijholt, I.M. Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus 2010, 20, 1018–1026. [Google Scholar] [CrossRef]
- Ouyang, M.; Zhang, L.; Zhu, J.J.; Schwede, F.; Thomas, S.A. Epac signaling is required for hippocampus-dependent memory retrieval. Proc. Natl. Acad. Sci. USA 2008, 105, 11993–11997. [Google Scholar] [CrossRef]
- Zhou, L.; Ma, S.L.; Yeung, P.K.K.; Wong, Y.H.; Tsim, K.W.K.; So, K.F.; Lam, L.C.W.; Chung, S.K. Anxiety and depression with neurogenesis defects in exchange protein directly activated by cAMP 2-deficient mice are ameliorated by a selective serotonin reuptake inhibitor, Prozac. Transl. Psychiatry 2016, 6, e881. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Patriarchi, T.; Buonarati, O.R.; Hell, J.W. Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by β2 adrenergic receptor/PKA and Ca 2+/CaMKII signaling. EMBO J. 2018, 37, e99771. [Google Scholar] [CrossRef]
- Diering, G.H.; Gustina, A.S.; Huganir, R.L. PKA-GluA1 Coupling via AKAP5 Controls AMPA Receptor Phosphorylation and Cell-Surface Targeting during Bidirectional Homeostatic Plasticity. Neuron 2014, 84, 790–805. [Google Scholar] [CrossRef]
- Sanderson, J.L.; Freund, R.K.; Gorski, J.A.; Dell’acqua, M.L. β-Amyloid disruption of LTP/LTD balance is mediated by AKAP150-anchored PKA and Calcineurin regulation of Ca2+-permeable AMPA receptors. Cell Rep. 2021, 37, 109786. [Google Scholar] [CrossRef]
- Jia, Z.; Collingridge, G.L. Learning about Synaptic GluA3. Neuron 2017, 93, 254–256. [Google Scholar] [CrossRef]
- Renner, M.C.; Albers, E.H.; Gutierrez-Castellanos, N.; Reinders, N.R.; van Huijstee, A.N.; Xiong, H.; Lodder, T.R.; Kessels, H.W. Synaptic plasticity through activation of GluA3-containing AMPA-receptors. Elife 2017, 6, 25462. [Google Scholar] [CrossRef] [PubMed]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange Protein Directly Activated by cAMP (epac): A Multidomain cAMP Mediator in the Regulation of Diverse Biological Functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed]
- Martín, R.; García-Font, N.; Suárez-Pinilla, A.S.; Bartolomé-Martín, D.; Ferrero, J.J.; Luján, R.; Torres, M.; Sánchez-Prieto, J. β-Adrenergic Receptors/Epac Signaling Increases the Size of the Readily Releasable Pool of Synaptic Vesicles Required for Parallel Fiber LTP. J. Neurosci. 2020, 40, 8604–8617. [Google Scholar] [CrossRef]
- Nijholt, I.M.; Dolga, A.M.; Ostroveanu, A.; Luiten, P.G.; Schmidt, M.; Eisel, U.L. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal. 2008, 20, 1715–1724. [Google Scholar] [CrossRef]
- Chen, X.; Garelick, M.G.; Wang, H.; Li, V.; Athos, J.; Storm, D.R. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat. Neurosci. 2005, 8, 925–931. [Google Scholar] [CrossRef]
- Kritman, M.; Maroun, M. Inhibition of the PI3 kinase cascade in corticolimbic circuit: Temporal and differential effects on contextual fear and extinction. Int. J. Neuropsychopharmacol. 2013, 16, 825–833. [Google Scholar] [CrossRef]
- Zanca, R.M.; Sanay, S.; Avila, J.A.; Rodriguez, E.; Shair, H.N.; Serrano, P.A. Contextual fear memory modulates PSD95 phosphorylation, AMPAr subunits, PKMζ and PI3K differentially between adult and juvenile rats. Neurobiol. Stress 2019, 10, 100139. [Google Scholar] [CrossRef]
- Man, H.-Y.; Wang, Q.; Lu, W.-Y.; Ju, W.; Ahmadian, G.; Liu, L.; D’Souza, S.; Wong, T.; Taghibiglou, C.; Lu, J.; et al. Activation of PI3-Kinase Is Required for AMPA Receptor Insertion during LTP of mEPSCs in Cultured Hippocampal Neurons. Neuron 2003, 38, 611–624. [Google Scholar] [CrossRef]
- Gelinas, J.N.; Banko, J.L.; Peters, M.M.; Klann, E.; Weeber, E.J.; Nguyen, P.V. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn. Mem. 2008, 15, 403–411. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Biou, V.; Xu, W.; Schlüter, O.; Malenka, R.C. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat. Neurosci. 2009, 12, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Gündüz, D.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Hamm, C.; Aslam, M. Role of PI3K/Akt and MEK/ERK Signalling in cAMP/Epac-Mediated Endothelial Barrier Stabilisation. Front. Physiol. 2019, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, C.; Feng, C.; Wu, Y.; Lu, H.; He, P.; Yang, X.; Zhu, F.; Qi, Q.; Gao, Y.; et al. Inhibition of phosphodiesterase-4 attenuates murine ulcerative colitis through interference with mucosal immunity. Br. J. Pharmacol. 2019, 176, 2209–2226. [Google Scholar] [CrossRef]
- Lin, S.L.; Johnson-Farley, N.N.; Lubinsky, D.R.; Cowen, D.S. Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J. Neurochem. 2003, 87, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Wang, M.; Morozov, Y.M.; Yang, S.; Mentone, S.; Zeiss, C.; Duque, A.; Rakic, P.; Horvath, T.L.; et al. Age-related calcium dysregulation linked with tau pathology and impaired cognition in non-human primates. Alzheimer’s Dement. 2021, 17, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Benitez, M.J.; Cuadros, R.; Jimenez, J.S. Phosphorylation and Dephosphorylation of Tau Protein by the Catalytic Subunit of PKA, as Probed by Electrophoretic Mobility Retard. J Alzheimers Dis 2021, 79, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Jicha, G.A.; Weaver, C.; Lane, E.; Vianna, C.; Kress, Y.; Rockwood, J.; Davies, P. cAMP-Dependent Protein Kinase Phosphorylations on Tau in Alzheimer’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 7486–7494. [Google Scholar] [CrossRef]
- Musheshe, N.; Oun, A.; Sabogal-Guáqueta, A.M.; Trombetta-Lima, M.; Mitchel, S.C.; Adzemovic, A.; Speek, O.; Morra, F.; van der Veen, C.H.J.T.; Lezoualc’h, F.; et al. Pharmacological Inhibition of Epac1 Averts Ferroptosis Cell Death by Preserving Mitochondrial Integrity. Antioxidants 2022, 11, 314. [Google Scholar] [CrossRef]
- Scheggia, D.; Stanic, J.; Italia, M.; La Greca, F.; Zianni, E.; Benussi, A.; Borroni, B.; Di Luca, M.; Gardoni, F. GluA3 autoantibodies induce alterations in dendritic spine and behavior in mice. Brain Behav. Immun. 2021, 97, 89–101. [Google Scholar] [CrossRef]
- Enache, D.; Pereira, J.B.; Jelic, V.; Winblad, B.; Nilsson, P.; Aarsland, D.; Bereczki, E. Increased Cerebrospinal Fluid Concentration of ZnT3 Is Associated with Cognitive Impairment in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1143–1155. [Google Scholar] [CrossRef]
- McPhee, I.; Gibson, L.; Kewney, J.; Darroch, C.; Stevens, P.; Spinks, D.; Cooreman, A.; MacKenzie, S. Cyclic nucleotide signalling: A molecular approach to drug discovery for Alzheimer’s disease. Biochem. Soc. Trans. 2005, 33, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level Neuronal Expression of Aβ1–42 in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Carrico, Z.; Alfonso, S.; Marino, M.; Koymans, K.; Kessels, H.W.; Malinow, R. PSD-95 protects synapses from β-amyloid. Cell Rep. 2021, 35, 109194. [Google Scholar] [CrossRef]
- Josselyn, S.A.; Tonegawa, S. Memory engrams: Recalling the past and imagining the future. Science 2020, 367, 4325. [Google Scholar] [CrossRef] [PubMed]
- Ostroveanu, A.; Van der Zee, E.A.; Dolga, A.M.; Luiten, P.G.; Eisel, U.L.; Nijholt, I.M. A-kinase anchoring protein 150 in the mouse brain is concentrated in areas involved in learning and memory. Brain Res. 2007, 1145, 97–107. [Google Scholar] [CrossRef]
- Mao, L.; Takamiya, K.; Thomas, G.; Lin, D.-T.; Huganir, R.L. GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 19038–19043. [Google Scholar] [CrossRef]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.-H.; Hortobágyi, T.; Winblad, B.; Ballard, C.; Lehtiö, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef]
- Sultana, R.; Banks, W.A.; Butterfield, D.A. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J. Neurosci. Res. 2009, 88, 469–477. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 354–385. [Google Scholar] [CrossRef]
- Chan, C.B.; Chen, Y.; Liu, X.; Tang, X.; Lee, C.W.; Mei, L.; Ye, K. PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression. EMBO J. 2011, 30, 4274–4286. [Google Scholar] [CrossRef]
- Zobon, N.T.M.; Jędrzejewska-Szmek, J.; Blackwell, K.T. Temporal pattern and synergy influence activity of ERK signaling pathways during L-LTP induction. Elife 2021, 10, 64644. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.M.; Park, S.J.; Lee, S.; Shin, C.Y.; Cheong, J.H.; Ryu, J.H. Hippocampal Extracellular Signal-Regulated Kinase Signaling has a Role in Passive Avoidance Memory Retrieval Induced by GABAA Receptor Modulation in Mice. Neuropsychopharmacology 2012, 37, 1234–1244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fukushima, H.; Zhang, Y.; Kida, S. Active Transition of Fear Memory Phase from Reconsolidation to Extinction through ERK-Mediated Prevention of Reconsolidation. J. Neurosci. 2021, 41, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Cone, R.D.; Scott, J.D. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J. Biol. Chem. 1992, 267, 16816–16823. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Schwede, F.; Bertinetti, D.; Langerijs, C.N.; Hadders, M.A.; Wienk, H.; Ellenbroek, J.H.; de Koning, E.J.P.; Bos, J.L.; Herberg, F.W.; Genieser, H.-G.; et al. Structure-Guided Design of Selective Epac1 and Epac2 Agonists. PLoS Biol. 2015, 13, e1002038. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Musheshe, N.; van der Veen, C.H.J.T.M.; Kessels, H.W.; Dolga, A.; De Deyn, P.; Eisel, U.; Schmidt, M. The Expression of Epac2 and GluA3 in an Alzheimer’s Disease Experimental Model and Postmortem Patient Samples. Biomedicines 2023, 11, 2096. https://doi.org/10.3390/biomedicines11082096
Zhang T, Musheshe N, van der Veen CHJTM, Kessels HW, Dolga A, De Deyn P, Eisel U, Schmidt M. The Expression of Epac2 and GluA3 in an Alzheimer’s Disease Experimental Model and Postmortem Patient Samples. Biomedicines. 2023; 11(8):2096. https://doi.org/10.3390/biomedicines11082096
Chicago/Turabian StyleZhang, Tong, Nshunge Musheshe, Christina H. J. T. M. van der Veen, Helmut W. Kessels, Amalia Dolga, Peter De Deyn, Ulrich Eisel, and Martina Schmidt. 2023. "The Expression of Epac2 and GluA3 in an Alzheimer’s Disease Experimental Model and Postmortem Patient Samples" Biomedicines 11, no. 8: 2096. https://doi.org/10.3390/biomedicines11082096
APA StyleZhang, T., Musheshe, N., van der Veen, C. H. J. T. M., Kessels, H. W., Dolga, A., De Deyn, P., Eisel, U., & Schmidt, M. (2023). The Expression of Epac2 and GluA3 in an Alzheimer’s Disease Experimental Model and Postmortem Patient Samples. Biomedicines, 11(8), 2096. https://doi.org/10.3390/biomedicines11082096