


Arsenic Inhibits Proliferation and Induces Autophagy of Tumor Cells in Pleural Effusion of Patients with Non-Small Cell Lung Cancer Expressing EGFR with or without Mutations via PI3K/AKT/mTOR Pathway
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects, Reagents, and Instruments
2.1.1. Subjects
2.1.2. Reagents and Instruments
2.2. Methods
2.2.1. Collection of Pleural Effusion and Intrapleural Administration
2.2.2. Tumor Cell Isolation from Pleural Effusion and Wright’s Staining
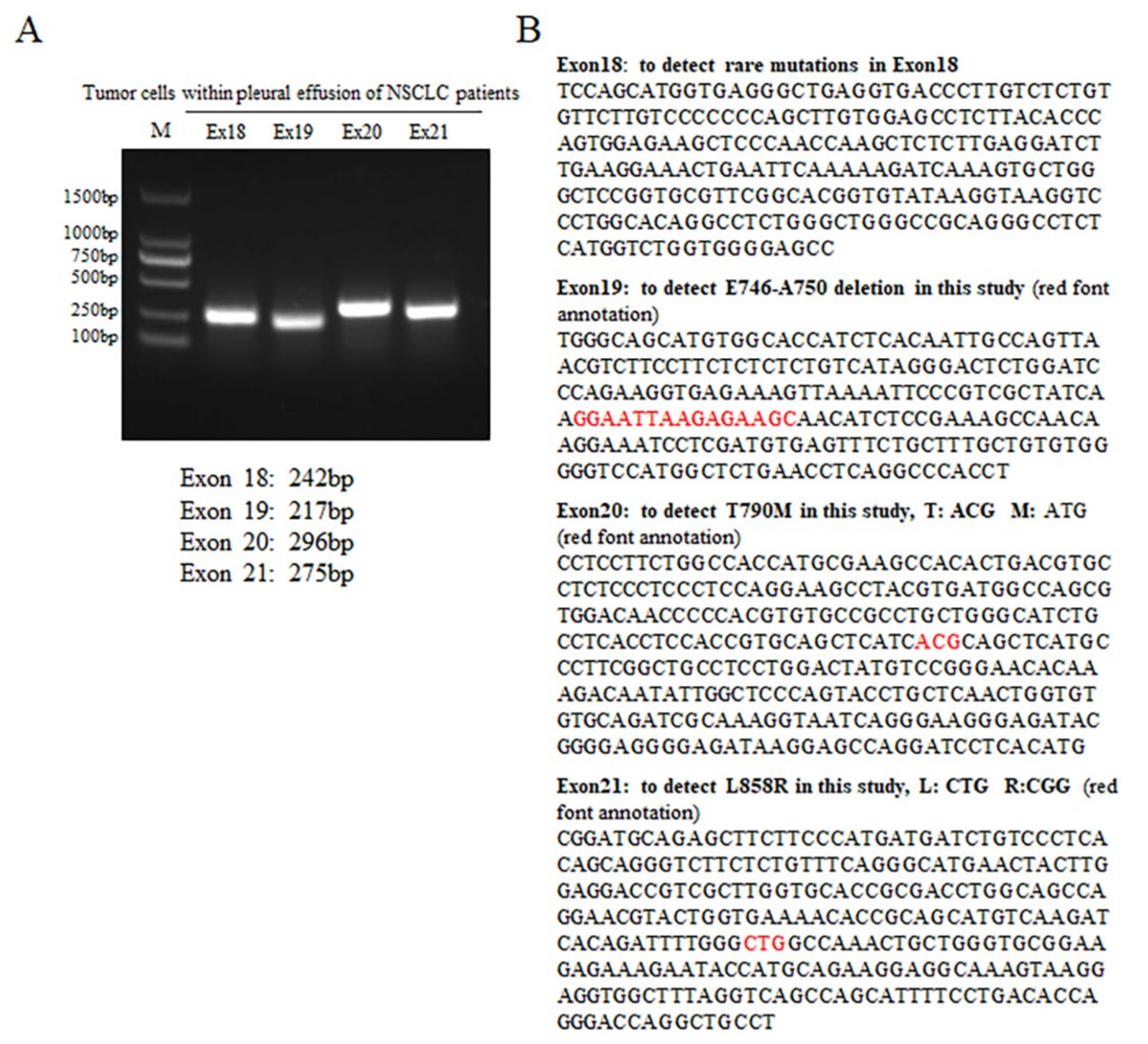
2.2.3. PCR and Sequencing
2.2.4. Cell Culture and Drug Treatment
2.2.5. WB Assay
2.2.6. IF Assay
2.2.7. Statistical Analysis
3. Results
3.1. Identification of the EGFR Genotype of Tumor Cells Derived from NSCLC Patients’ Pleural Effusion
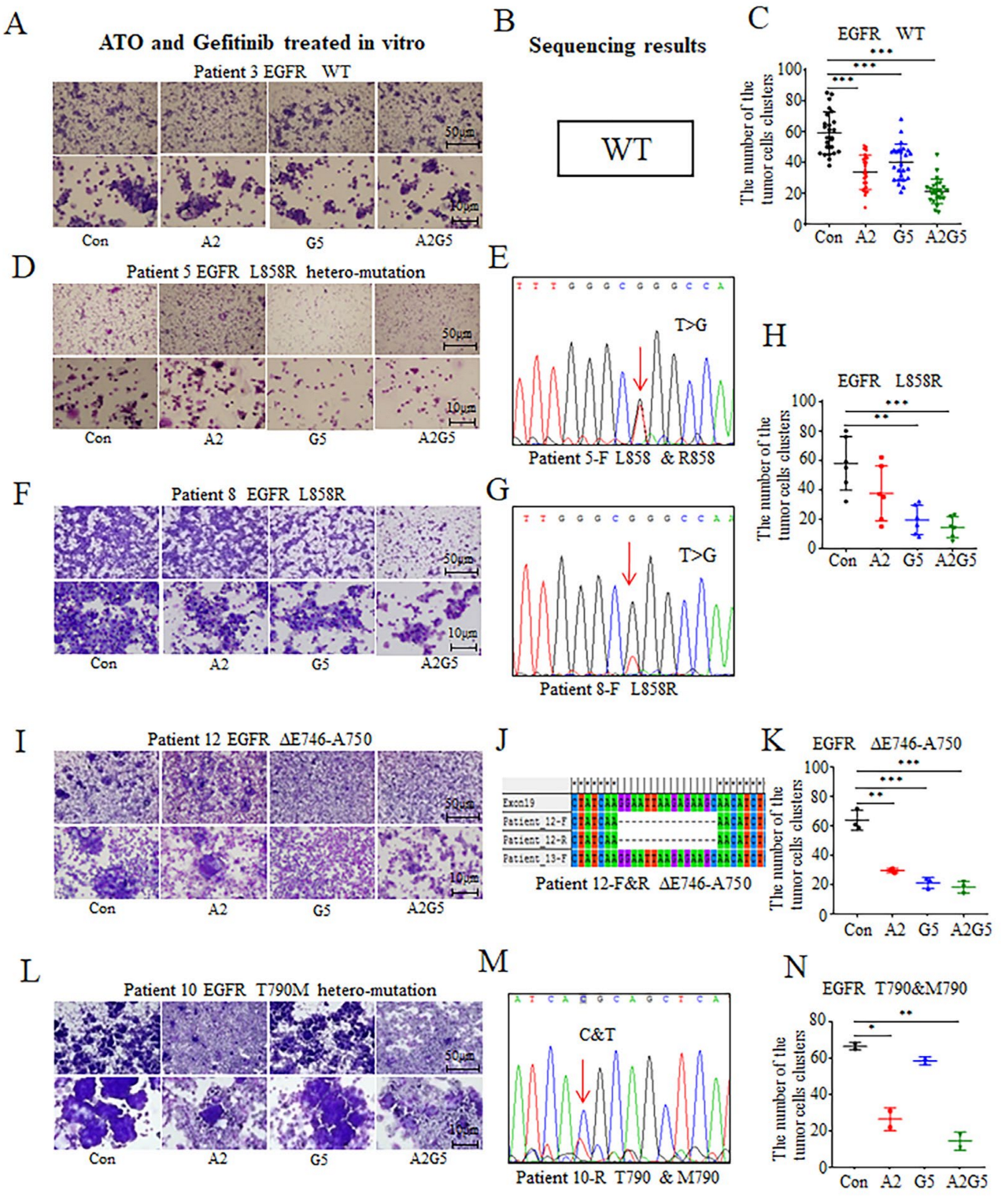
3.2. In Vitro Effects of Arsenic and Gefitinib on Primary Tumor Cells Derived from NSCLC Patients’ Pleural Effusion
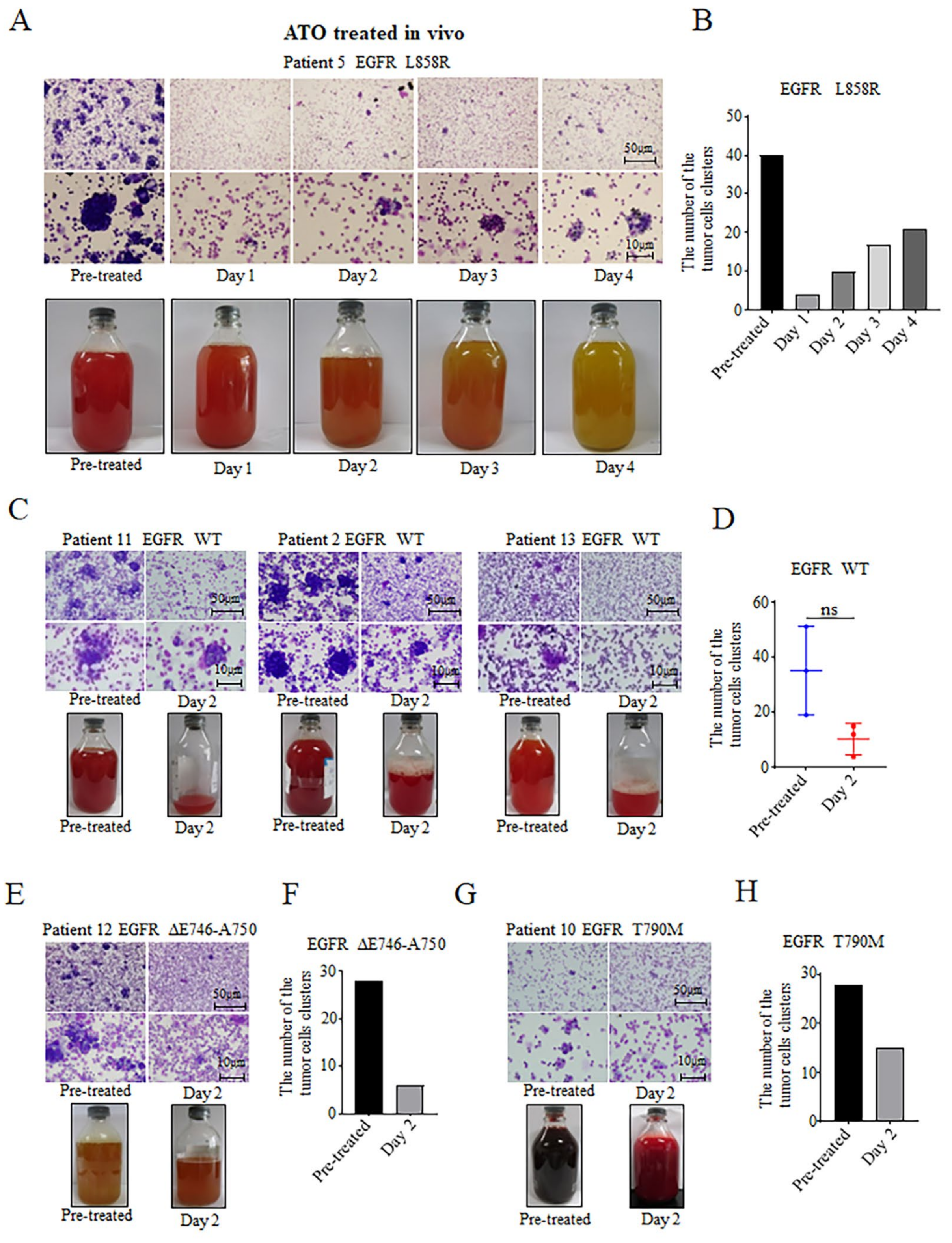
3.3. Inhibitory Effects of ATO Intrapleural Administration on NSCLC Tumor Cells
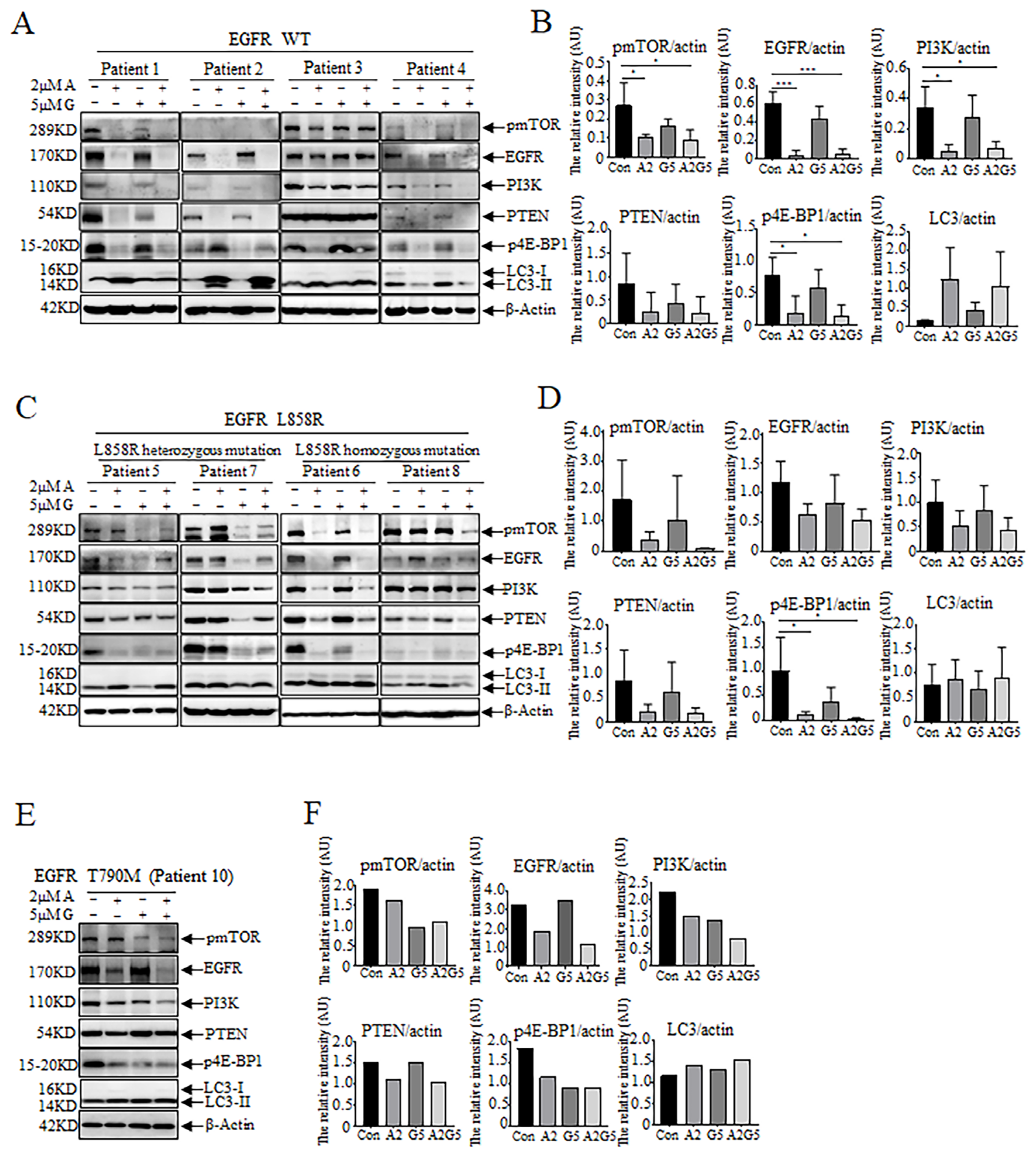
3.4. Effects of Arsenic and Gefitinib on Proliferation and Autophagy in Tumor Cells in Pleural Effusion
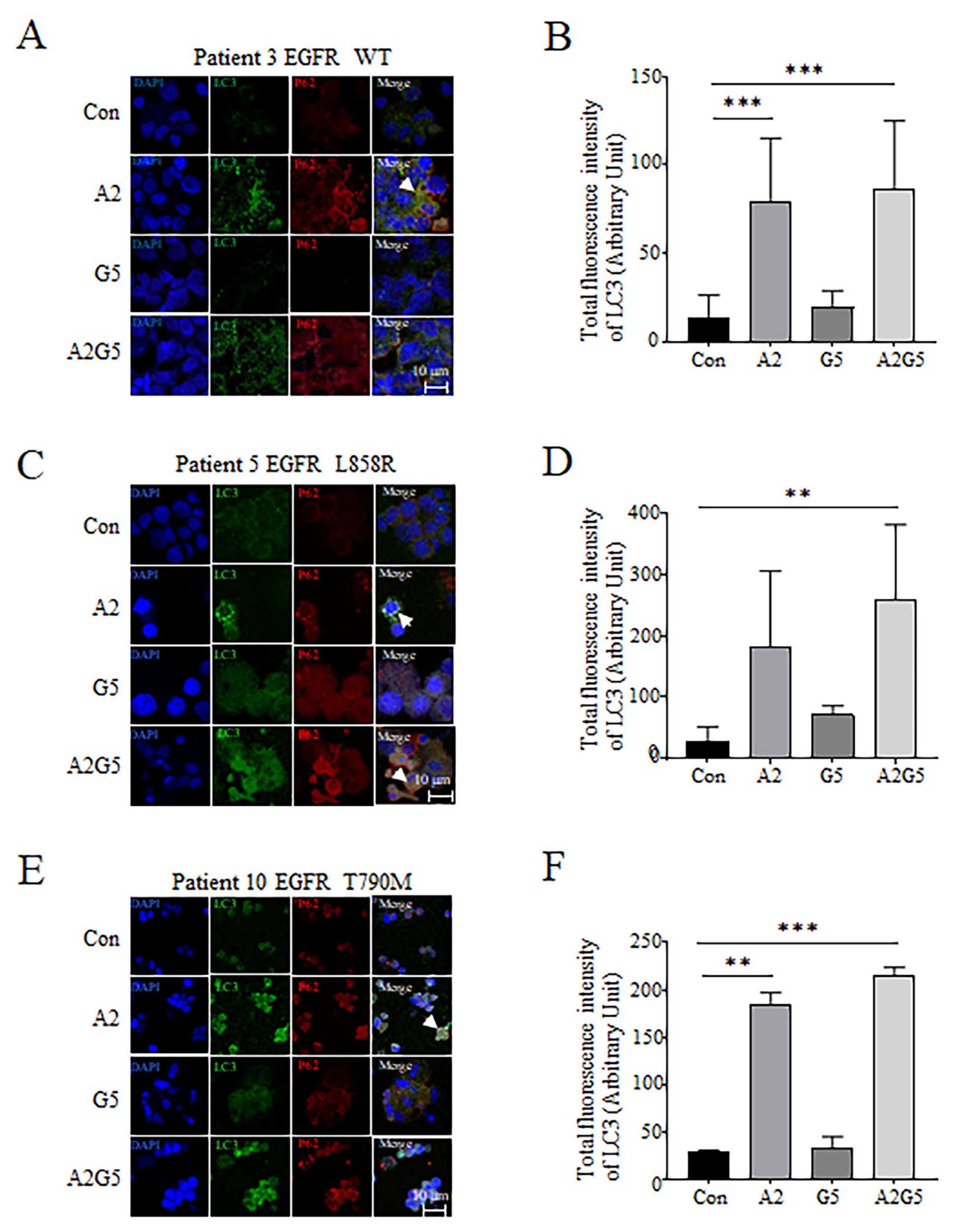
3.5. Detection of LC3 and P62 Expression and Distribution in Tumor Cells Derived from Pleural Effusion Treated with Arsenic and Gefitinib by IF
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Yang, S.-R.; Schultheis, A.M.; Yu, H.; Mandelker, D.; Ladanyi, M.; Büttner, R. Precision medicine in non-small cell lung cancer: Current applications and future directions. Semin. Cancer Biol. 2022, 84, 184–198. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.-S.; Squire, J.; et al. Molecular Testing Guideline for Selection of Lung Cancer Patients for EGFR and ALK Tyrosine Kinase Inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2013, 137, 828–860. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular testing guideline for selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: Guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. FDA Approval of Gefitinib for the Treatment of Patients with Metastatic EGFR Mutation–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef]
- Kohsaka, S.; Petronczki, M.; Solca, F.; Maemondo, M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Futur. Oncol. 2019, 15, 637–652. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, H.-M.; Li, Y.; Liu, Q.-F.; Hu, Y.; Zhou, J.-F.; Jin, J.; Hu, J.-D.; Liu, T.; Wu, D.-P.; et al. Arsenic trioxide replacing or reducing chemotherapy in consolidation therapy for acute promyelocytic leukemia (APL2012 trial). Proc. Natl. Acad. Sci. USA 2021, 118, e2020382118. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.-F.; Wu, C.-F.; Xu, F.; Shen, Z.-X.; Zhu, Y.-M.; Li, J.-M.; Tang, W.; Zhao, W.-L.; Wu, W.; et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3342–3347. [Google Scholar] [CrossRef]
- Mao, J.; Ma, L.; Shen, Y.; Zhu, K.; Zhang, R.; Xi, W.; Ruan, Z.; Luo, C.; Chen, Z.; Xi, X.; et al. Arsenic circumvents the gefitinib resistance by binding to P62 and mediating autophagic degradation of EGFR in non-small cell lung cancer. Cell Death Dis. 2018, 9, 963. [Google Scholar] [CrossRef]
- Tian, W.; Sun, Y.; Cheng, Y.; Ma, X.; Du, W.; Shi, W.; Guo, Q. Arsenic sulfide reverses cisplatin resistance in non-small cell lung cancer in vitro and in vivo through targeting PD-L1. Thorac. Cancer 2021, 12, 2551–2563. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Deng, X.; He, W.; Yang, Q.; Ni, L.; Shasaltaneh, M.D.; Maghsoudloo, M.; Yang, G.; Wu, J.; Imani, S.; et al. Treatment of malignant pleural effusion in non-small cell lung cancer with VEGF-directed therapy. Ann. Med. 2022, 54, 1357–1371. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, D.; Huang, H.; Xiu, Q. Clinical value of arsenic in treating lung cancer complicated with pleural effusion. China Oncol. 2006, 16, 681–682. [Google Scholar]
- Shi, X.; Yang, M.; Huang, H.; Fang, Z.; Shi, Z.; Tang, H.; Wang, X.; Chen, Y.; Lü, Y.; Chen, D.; et al. Efficacy and mechanism of arsenic trioxide intrapleural injection in non-small cell lung cancer patients with malignant pleural effusions. J. Intern. Med. Concepts Pract. 2019, 14, 77–82. [Google Scholar]
- Chen, G.Q.; Shi, X.G.; Tang, W.; Xiong, S.M.; Zhu, J.; Cai, X.; Han, Z.G.; Ni, J.H.; Shi, G.Y.; Jia, P.M.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997, 89, 3345–3353. [Google Scholar]
- Zhang, Q.-Y.; Mao, J.-H.; Liu, P.; Huang, Q.-H.; Lu, J.; Xie, Y.-Y.; Weng, L.; Zhang, Y.; Chen, Q.; Chen, S.-J.; et al. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3378–3383. [Google Scholar] [CrossRef]
- Huang, Z.-Y.; Shao, M.-M.; Zhang, J.-C.; Yi, F.-S.; Du, J.; Zhou, Q.; Wu, F.-Y.; Li, S.; Li, W.; Huang, X.-Z.; et al. Single-cell analysis of diverse immune phenotypes in malignant pleural effusion. Nat. Commun. 2021, 12, 6690. [Google Scholar] [CrossRef]
- Psallidas, I.; Kalomenidis, I.; Porcel, J.M.; Robinson, B.W.; Stathopoulos, G. Malignant pleural effusion: From bench to bedside. Eur. Respir. Rev. 2016, 25, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, H.; Shi, X.; Huo, Z.; Zhang, J.; Liang, Z. Comparison of Next-Generation Sequencing, Quantitative PCR, and Sanger Sequencing for Mutation Profiling of EGFR, KRAS, PIK3CA and BRAF in Clinical Lung Tumors. Clin. Lab. 2016, 62, 689–696. [Google Scholar] [CrossRef]
- Xie, S.-L.; Yang, M.-H.; Chen, K.; Huang, H.; Zhao, X.-W.; Zang, Y.-S.; She-Ling, X. Efficacy of Arsenic Trioxide in the Treatment of Malignant Pleural Effusion Caused by Pleural Metastasis of Lung Cancer. Cell Biochem. Biophys. 2015, 71, 1325–1333. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yan, X.J.; Zhou, Z.R.; Yang, F.F.; Wu, Z.Y.; Sun, H.B.; Liang, W.X.; Song, A.X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- Chen, S.-J.; Zhou, G.-B.; Zhang, X.-W.; Mao, J.-H.; De Thé, H.; Chen, Z. From an old remedy to a magic bullet: Molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood 2011, 117, 6425–6437. [Google Scholar] [CrossRef]
- Mao, J.H.; Sun, X.Y.; Liu, J.X.; Zhang, Q.Y.; Liu, P.; Huang, Q.H.; Li, K.K.; Chen, Q.; Chen, Z.; Chen, S.J. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 21683–21688. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Huang, W.; Liu, X.; Liu, X.; Chen, N.; Xu, Q.; Hu, Y.; Song, W.; Zhou, J. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J. Exp. Clin. Cancer Res. 2018, 37, 304. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zang, H.; Zheng, H.; Zhan, Y.; Yang, Y.; Zhang, Y.; Liu, S.; Feng, J.; Wen, Q.; Long, M.; et al. Overexpression of p-Akt, p-mTOR and p-eIF4E proteins associates with metastasis and unfavorable prognosis in non-small cell lung cancer. PLoS ONE 2020, 15, e0227768. [Google Scholar] [CrossRef] [PubMed]
- Crees, Z.D.; Shearrow, C.; Lin, L.; Girard, J.; Arasi, K.; Bhoraskar, A.; Berei, J.; Eckburg, A.; Anderson, A.D.; Garcia, C.; et al. EGFR/c-Met and mTOR signaling are predictors of survival in non-small cell lung cancer. Ther. Adv. Med Oncol. 2020, 12, 1758835920953731. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2011, 19, 87–95. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Fang, S.; Wan, X.; Zou, X.; Sun, S.; Hao, X.; Liang, C.; Zhang, Z.; Zhang, F.; Sun, B.; Li, H.; et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 2021, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/mTOR Pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Lim, J.U.; Yeo, C.D. Update on adjuvant therapy in completely resected NSCLC patients. Thorac. Cancer 2022, 13, 277–283. [Google Scholar] [CrossRef]
- Zhong, J.; Bai, H.; Wang, Z.; Duan, J.; Zhuang, W.; Wang, D.; Wan, R.; Xu, J.; Fei, K.; Ma, Z.; et al. Treatment of advanced non-small cell lung cancer with driver mutations: Current applications and future directions. Front. Med. 2023, 17, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Mamdani, H.; Matosevic, S.; Khalid, A.B.; Durm, G.; Jalal, S.I. Immunotherapy in Lung Cancer: Current Landscape and Future Directions. Front. Immunol. 2022, 13, 823618. [Google Scholar] [CrossRef]
- Yoneda, K.; Imanishi, N.; Ichiki, Y.; Tanaka, F. Treatment of Non-small Cell Lung Cancer with EGFR-mutations. J. UOEH 2019, 41, 153–163. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Tan, C.-S.; Kumarakulasinghe, N.B.; Huang, Y.-Q.; Ang, Y.L.E.; Choo, J.R.-E.; Goh, B.-C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Yang, C.-H.; Huang, C.-J.; Yang, C.-S.; Chu, Y.-C.; Cheng, A.-L.; Whang-Peng, J.; Yang, P.-C. Gefitinib Reverses Chemotherapy Resistance in Gefitinib-Insensitive Multidrug Resistant Cancer Cells Expressing ATP-Binding Cassette Family Protein. Cancer Res. 2005, 65, 6943–6949. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Zeng, L.; Xu, M.; Wang, X.Q.; Yang, D.H.; Chen, Z.S. Osimertinib (AZD9291), a mutant-selective EGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Molecules 2016, 21, 1236. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, J.; Kryczka, J.; Czarnecka-Chrebelska, K.H.; Brzeziańska-Lasota, E. Molecular mechanisms of chemoresistance induced by cisplatin in NSCLC cancer therapy. Int. J. Mol. Sci. 2021, 22, 8885. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, K.; Imanishi, N.; Ichiki, Y.; Tanaka, F. Immune Checkpoint Inhibitors (ICIs) in Non-Small Cell Lung Cancer (NSCLC). J. UOEH 2018, 40, 173–189. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ishibashi, K.; Yumoto, A.; Nishio, K.; Ogasawara, Y. Utilization of arsenic trioxide as a treatment of cisplatin-resistant non-small cell lung cancer PC-9/CDDP and PC-14/CDDP cells. Oncol. Lett. 2015, 10, 805–809. [Google Scholar] [CrossRef]
- Li, H.; Zhu, X.; Zhang, Y.; Xiang, J.; Chen, H. Arsenic trioxide exerts synergistic effects with cisplatin on non-small cell lung cancer cells via apoptosis induction. J. Exp. Clin. Cancer Res. 2009, 28, 110. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.P.; Xiu, Q.Y.; Li, B.; Liu, Y.A.; Zhang, L.Z. Arsenic trioxide inhibits the growth of human lung cancer cell lines via cell cycle arrest and induction of apoptosis at both normoxia and hypoxia. Toxicol. Ind. Health 2009, 25, 505–515. [Google Scholar]
- Chang, K.-J.; Yang, M.-H.; Zheng, J.-C.; Li, B.; Nie, W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. Am. J. Transl. Res. 2016, 8, 1133–1143. [Google Scholar]
- Vizzardi, E.; Zanini, G.; Antonioli, E.; D’aloia, A.; Raddino, R.; Cas, L.D. QT Prolongation: A Case of Arsenical Pericardial and Pleural Effusion. Cardiovasc. Toxicol. 2007, 8, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nagai, S.; Miyashita, S.-I.; Kaise, T.; Ichikawa, M.; Kumano, K.; Hangaishi, A.; Nannya, Y.; Kurokawa, M. Arsenic-induced pericardial and pleural effusion without acute promyelocytic leukemia differentiation syndrome. Leuk. Res. 2010, 34, e25–e26. [Google Scholar] [CrossRef]
- Tandberg, D.J.; Tong, B.C.; Ackerson, B.G.; Kelsey, C.R. Surgery versus stereotactic body radiation therapy for stage I non-small cell lung cancer: A comprehensive review. Cancer 2018, 124, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of radiosensitizers in caner radiotherapy. Int. J. Nanomed. 2021, 16, 1083–1102. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the dynamic tumor microenvironment in controversies regarding immune checkpoint inhibitors for the treatment of non-small cell lung cancer (NSCLC) with EGFR mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, M.; Filetti, M.; Occhipinti, M.; Marinelli, D.; Scalera, S.; Terrenato, I.; Sperati, F.; Pallocca, M.; Rizzo, F.; Gelibter, A.; et al. Efficacy of immunotherapy in lung cancer with co-occurring mutations in NOTCH and homologous repair genes. J. Immunother. Cancer 2020, 8, e000946. [Google Scholar] [CrossRef]
- He, T.; Cao, J.; Xu, J.; Lv, W.; Hu, J. Minimally invasive therapies for early stage non-small cell lung cancer. Zhongguo Fei Ai Za Zhi 2020, 23, 479–486. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Character (n = 36) | Number (%) |
|---|---|
| Gender (%) | |
| Female | 13 (36.1) |
| Male | 23 (63.9) |
| Age (median [Q1; Q3]) | 63.5 [54.0; 73.0] |
| Smoking (%) | |
| Never | 19 (52.8) |
| Former | 3 (8.33) |
| Current | 14 (38.9) |
| Type (%) | |
| Adeno-carcinoma | 32 (88.9) |
| Poorly differentiated adeno-carcinoma | 4 (11.1) |
| T stage (%) | |
| T1 | 2 (5.56) |
| T2 | 9 (25.0) |
| T3 | 8 (22.2) |
| T4 | 17 (47.2) |
| N stage (%) | |
| N1 | 6 (16.7) |
| N2 | 12 (33.3) |
| N3 | 18 (50.0) |
| M stage (%) | |
| M1a | 17 (47.2) |
| M1b | 16 (44.4) |
| M1c | 3 (8.33) |
| Appearance of pleural effusions (%) | |
| Yellow and muddy | 17 (47.2) |
| Bloody and muddy | 19 (52.8) |
| EGFR Genotype (%) | |
| WT | 25 (69.4) |
| L858R | 6 (16.7) |
| T790M | 2 (5.56) |
| ΔE746-A750 | 3 (8.33) |
| Therapies | Efficacy | Mechanisms | Side Effects | |||||
|---|---|---|---|---|---|---|---|---|
| EGFR WT | EGFR L858R or ΔE746-A750 | EGFR T790M | PD-1/PD-L1 | Chemo- Resistance | PFS/OR/OS | |||
| TKIs | ||||||||
| Gefitinib | − [51,52] | ++ [51,52] | − [51,52] | +/− [56] | OR 71.2% in NSCLC with EGFR mutations [51], 12-month PFS 24.9% [52]. | Inhibition of the EGFR tyrosine kinase activity by binding to ATP pocket [16,51]. | Rash or acne, diarrhea, dry skin, anorexia, pruritus, stomatitis, asthenic conditions, nausea, paronychia, vomiting, constipation, neurotoxic effects, myalgia, arthralgia [52] | |
| Osimertinib | − [54] | + [54] | +++ [51,53,54] | +/− [57] | PFS 10.1 months [14,51], PFS 18.9 months in a first-line treatment in the FLAURA trail [51]. | Selective inhibition mutant EGFR through irreversible and covalent binding to C797 in the tyrosine kinase domain [51,54]. | Diarrhea, rash, dry skin, paronychia [14]. | |
| Chemotherapy | ||||||||
| Cisplatin/ Pemetrexed | +/− [58,59] | +/− [51,52] | +/− [58,59] | − [58,59] | PFS 4.4 months [14,51]. | Formation of covalent adducts between platinum complexes and DNA, RNA, and proteins [58,59]. | Nausea, decreased appetite, constipation, anemia [14]. | |
| Carboplatin /Paclitaxel | +/− [51,52] | +/− [51,52] | +/− [51,52] | − [58,59] | 12-month PFS 6.7% [52]. | Formation of covalent adducts between platinum complexes and DNA, RNA, and proteins [58,59]. | Rash or acne, diarrhea, dry skin, anorexia, pruritus, stomatitis, asthenic conditions, nausea, paronychia, vomiting, constipation, neurotoxic effects, myalgia, arthralgia [52]. | |
| ICIs | ||||||||
| Nivolumab (anti-PD-1) | +/− [60] | +/− [60] | +/− [60] | ++ [60] | + [61] | OR 20%, PFS 3.5 months, OS 9.2 months [60]. | Restore the ability of cancer immunity to kill tumor cells by blocking PD-1 [60]. | Pneumonitis, hepatitis, neurotoxic effects, myocarditis, toxicity-related fatality rates is 0.36% [62]. |
| Pembrolizumab (anti-PD-1) | +/− [60] | +/− [60] | +/− [60] | ++ [60] | + [61] | OR 18%, PFS 3.9 months, OS 10.4 months [60]. | Restore the ability of cancer immunity to kill tumor cells by blocking PD-1 [60]. | Pneumonitis, hepatitis, neurotoxic effects, myocarditis, toxicity-related fatality rates is 0.36% [62]. |
| Durvalumab (anti-PD-L1) | +/− [60] | +/− [60] | +/− [60] | ++ [9,50] | + [9,50] | Median PFS 16.8 months [9], 12-month PFS 55.9%, 18-months PFS 44.2 [9]. | Restore the ability of cancer immunity to kill tumor cells by blocking PD-L1 [60]. | Diarrhea, pneumonitis, rash, pruritus, cough, fatigue, dyspnea, pyrexia, decreased appetite, nausea, arthralgia, constipation, anemia [9]. |
| Atezolizumab (anti-PD-L1) | +/− [60] | +/− [60] | +/− [60] | ++ [60] | + [61] | OR 14.6%, PFS 2.7 months, OS 12.6 months [60]. | Restore the ability of cancer immunity to kill tumor cells by blocking PD-L1 [60]. | Pneumonitis, hepatitis, neurotoxic effects, myocarditis, toxicity-related fatality rates is 0.38% [62]. |
| Arsenic | ||||||||
| ATO | + [this study] | + [21,this study] | ++ [21,this study] | + [63] | 5-year DFS > 95% in APL [19] To treat pleural effusion in NSCLC, OR 70.8%, CR 8.3%, PR 62.5% [25]. | Degradation of the oncoproteins [21]; inhibition proliferation [21,64]; induction apoptosis [64] and autophagy [21]; cell cycle arrest [65]; reduction pleural vascular permeability [31]; inhibition of the cancer stem-like cells [66]; reversion cisplatin resistance [63]. | Tolerable and reversible grade I-II liver dysfunction [20]; reversible pleural and/or pericardial effusion [67,68]; mild gastrointestinal reactions, fever, chest pain, leukopenia [24,25]. | |
| Arsenic sulfide | + [22] | + [22] | Sensitize NSCLC cells to cisplatin through targeting PD-L1 [22]. | |||||
| Surgery | ||||||||
| Lobectomy Wedge resection Segmentectomy | The standard treatment for patients with stage I NSCLC [69]. | 3-year OR 82%, 5-year OR 66% [69]. | Lobectomy, wedge resection, segmentectomy can remove the lesion [69]. | Recurrence, pneumonia, respiratory failure, myocardial infarction [69]. | ||||
| Radiotherapy | ||||||||
| SBRT | SBRT is recommended for those who are not medically fit for surgery. SBRT is extremely well tolerated. SBRT is an outpatient procedure [69]. | 2-year OR 84%, 3-year OR 77%, 5-year OR 55.7%, 7-year OR 47.5% [69], Local tumor control rates > 90% [69]. | Induce DNA damage, terminate cell division and proliferation, lead to cell necrosis and apoptosis. Induce the generation of ROS, which can induce cellular stress in, and injure biomolecules, alter cellular signaling pathways [70]. | Mild fatigue, exacerbate degenerative arthritis in the shoulders, back, hips. Decreased pulmonary function. Chest pain, rib fractures. Esophagitis, skin irritation, brachial plexopathy [69]. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, J.; Shi, X.; Hua, L.; Yang, M.; Shen, Y.; Ruan, Z.; Li, B.; Xi, X. Arsenic Inhibits Proliferation and Induces Autophagy of Tumor Cells in Pleural Effusion of Patients with Non-Small Cell Lung Cancer Expressing EGFR with or without Mutations via PI3K/AKT/mTOR Pathway. Biomedicines 2023, 11, 1721. https://doi.org/10.3390/biomedicines11061721
Mao J, Shi X, Hua L, Yang M, Shen Y, Ruan Z, Li B, Xi X. Arsenic Inhibits Proliferation and Induces Autophagy of Tumor Cells in Pleural Effusion of Patients with Non-Small Cell Lung Cancer Expressing EGFR with or without Mutations via PI3K/AKT/mTOR Pathway. Biomedicines. 2023; 11(6):1721. https://doi.org/10.3390/biomedicines11061721
Chicago/Turabian StyleMao, Jianhua, Xiaoqian Shi, Li Hua, Menghang Yang, Yan Shen, Zheng Ruan, Bing Li, and Xiaodong Xi. 2023. "Arsenic Inhibits Proliferation and Induces Autophagy of Tumor Cells in Pleural Effusion of Patients with Non-Small Cell Lung Cancer Expressing EGFR with or without Mutations via PI3K/AKT/mTOR Pathway" Biomedicines 11, no. 6: 1721. https://doi.org/10.3390/biomedicines11061721
APA StyleMao, J., Shi, X., Hua, L., Yang, M., Shen, Y., Ruan, Z., Li, B., & Xi, X. (2023). Arsenic Inhibits Proliferation and Induces Autophagy of Tumor Cells in Pleural Effusion of Patients with Non-Small Cell Lung Cancer Expressing EGFR with or without Mutations via PI3K/AKT/mTOR Pathway. Biomedicines, 11(6), 1721. https://doi.org/10.3390/biomedicines11061721