
Human In Vitro Skin Models for Wound Healing and Wound Healing Disorders

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
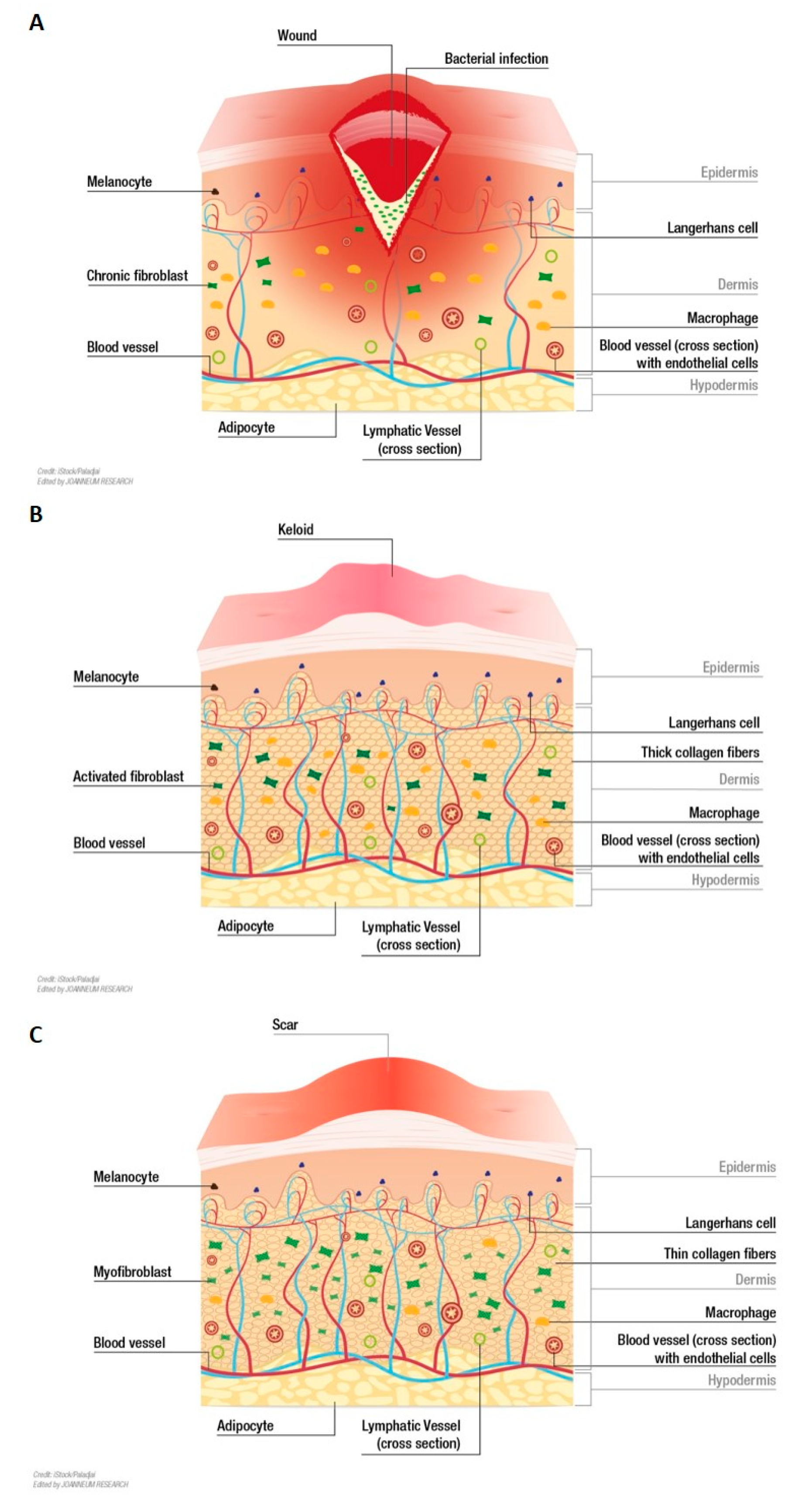
2. In Vitro Models for Wound Healing
3. In Vitro Models for Chronic Wounds
4. In Vitro Models for Excessive Scarring
4.1. Keloids
4.2. Hypertrophic Scars
5. Limitations and Future Perspectives of In Vitro Models for Wound Healing Disorders
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes: Cellular Mechanisms of Wound Repair. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef]
- Adib, Y.; Bensussan, A.; Michel, L. Cutaneous Wound Healing: A Review about Innate Immune Response and Current Therapeutic Applications. Mediat. Inflamm. 2022, 2022. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Murray, P.J.; Pearce, E.J. Metabolic orchestration of the wound healing response. Cell Metab. 2021, 33, 1726–1743. [Google Scholar] [CrossRef]
- Willenborg, S.; Injarabian, L.; Eming, S.A. Role of Macrophages in Wound Healing. Cold Spring Harb. Perspect. Biol. 2022, 14, a041216. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruber, J. Age-related aspects of cutaneous wound healing: A mini-review. Gerontology 2013, 59, 159–164. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef]
- Tarusha, L.; Paoletti, S.; Travan, A.; Marsich, E. Alginate membranes loaded with hyaluronic acid and silver nanoparticles to foster tissue healing and to control bacterial contamination of non-healing wounds. J. Mater. Sci. Mater. Med. 2018, 29, 22. [Google Scholar] [CrossRef]
- Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Kassymbek, K.; Jimi, S.; Saparov, A. Immunology of Acute and Chronic Wound Healing. Biomolecules 2021, 11, 700. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, H.; Wu, Z.; Chen, W.; Zhang, G. Identification of the potential targets for keloid and hypertrophic scar prevention. J. Dermatolog. Treat. 2018, 29, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Avci, P.; Sadasivam, M.; Gupta, A.; Melo, W.; Huang, Y.-Y.; Yin, R.; Chandran, R.; Kumar, R.; Otufowora, A.; Nyame, T.; et al. Animal models of skin disease for drug discovery. Expert Opin. Drug Discov. 2013, 8, 331–355. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.C.; Maibach, H.I. Animal models for percutaneous absorption. J. Appl. Toxicol. 2015, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dellambra, E.; Odorisio, T.; D’Arcangelo, D.; Failla, C.M.; Facchiano, A. Non-animal models in dermatological research. ALTEX 2019, 36, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Naldaiz-Gastesi, N.; Bahri, O.A.; Opez De Munain, A.L.; Mccullagh, K.J.A.; Izeta, A. The panniculus carnosus muscle: An evolutionary enigma at the intersection of distinct research fields. J. Anat. 2018, 233, 275–288. [Google Scholar] [CrossRef]
- Abdullahi, A.; Amini-Nik, S.; Jeschke, M.G. Animal models in burn research. Cell. Mol. Life Sci. 2014, 71, 3241–3255. [Google Scholar] [CrossRef]
- Gottrup, F.; Ågren, M.S.; Karlsmark, T. Models for use in wound healing research: A survey focusing on in vitro and in vivo adult soft tissue. Wound Repair Regen. 2000, 8, 83–96. [Google Scholar] [CrossRef]
- Dahiya, P. Burns as a model of SIRS. Front. Biosci. 2009, 14, 4962–4967. [Google Scholar] [CrossRef]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 2011, 969618. [Google Scholar] [CrossRef]
- Lorenz, H.P.; Longaker, M.T. Wounds: Biology, pathology, and management. Surg. Basic Sci. Clin. Evid. 2008, 191–208. [Google Scholar] [CrossRef]
- Pavletic, M.M. Atlas of Small Animal Wound Management and Reconstructive Surgery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Sullivan, T.P.; Eaglstein, W.H.; Davis, S.C.; Mertz, P. The pig as a model for human wound healing. Wound Repair Regen. 2001, 9, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Middelkoop, E.; Van Den Bogaerdt, A.J.; Lamme, E.N.; Hoekstra, M.J.; Brandsma, K.; Ulrich, M.M.W. Porcine wound models for skin substitution and burn treatment. Biomaterials 2004, 25, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Summerfield, A.; Meurens, F.; Ricklin, M.E. The immunology of the porcine skin and its value as a model for human skin. Mol. Immunol. 2015, 66, 14–21. [Google Scholar] [CrossRef]
- Niehues, H.; Bouwstra, J.A.; El Ghalbzouri, A.; Brandner, J.M.; Zeeuwen, P.L.J.M.; van den Bogaard, E.H. 3D skin models for 3R research: The potential of 3D reconstructed skin models to study skin barrier function. Exp. Dermatol. 2018, 27, 501–511. [Google Scholar] [CrossRef]
- Mathes, S.H.; Ruffner, H.; Graf-Hausner, U. The use of skin models in drug development. Adv. Drug Deliv. Rev. 2014, 69, 81–102. [Google Scholar] [CrossRef]
- Stamm, A.; Reimers, K.; Strauß, S.; Vogt, P.; Scheper, T.; Pepelanova, I. In vitro wound healing assays-state of the art. BioNanoMat 2016, 17, 79–87. [Google Scholar] [CrossRef]
- Groeber, F.; Holeiter, M.; Hampel, M.; Hinderer, S.; Schenke-Layland, K. Skin tissue engineering-In vivo and in vitro applications. Adv. Drug Deliv. Rev. 2011, 63, 352–366. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Pinto, B.I.; Tabor, A.J.; Stearns, D.M.; Diller, R.B.; Kellar, R.S. A Bench-Top In Vitro Wound Assay to Demonstrate the Effects of Platelet-Rich Plasma and Depleted Uranium on Dermal Fibroblast Migration. Appl. Vitr. Toxicol. 2016, 2, 151–156. [Google Scholar] [CrossRef]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res.—Rev. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adhes. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Riis, S.; Newman, R.; Ipek, H.; Andersen, J.I.; Kuninger, D.; Boucher, S.; Vemuri, M.C.; Pennisi, C.P.; Zachar, V.; Fink, T. Hypoxia enhances the woundhealing potential of adipose-derived stem cells in a novel human primary keratinocyte-based scratch assay. Int. J. Mol. Med. 2017, 39, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.Y.K.; Leung, E.P.Y.; Mak, N.K.; Wong, R.N.S. A Simplified Method for Quantifying Cell Migration/Wound Healing in 96-Well Plates. J. Biomol. Screen. 2010, 15, 427–433. [Google Scholar] [CrossRef]
- Giaever, I.; Keese, C.R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. USA 1991, 88, 7896–7900. [Google Scholar] [CrossRef]
- Anwer, S.; Szászi, K. Measuring Cell Growth and Junction Development in Epithelial Cells Using Electric Cell-Substrate Impedance Sensing (ECIS). Bio-Protocol 2020, 10, e3729. [Google Scholar] [CrossRef]
- Keese, C.R.; Wegener, J.; Walker, S.R.; Giaever, I. Electrical wound-healing assay for cells in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 1554–1559. [Google Scholar] [CrossRef]
- Hundsberger, H.; Koppensteiner, A.; Hofmann, E.; Ripper, D.; Pflüger, M.; Stadlmann, V.; Klein, C.T.; Kreiseder, B.; Katzlinger, M.; Eger, A.; et al. A Screening Approach for Identifying Gliadin Neutralizing Antibodies on Epithelial Intestinal Caco-2 Cells. SLAS Discov. Adv. Life Sci. R D 2017, 22, 1035–1043. [Google Scholar] [CrossRef]
- Pflüger, M.; Kapuscik, A.; Lucas, R.; Koppensteiner, A.; Katzlinger, M.; Jokela, J.; Eger, A.; Jacobi, N.; Wiesner, C.; Hofmann, E.; et al. A Combined Impedance and AlphaLISA-Based Approach to Identify Anti-inflammatory and Barrier-Protective Compounds in Human Endothelium. J. Biomol. Screen. 2013, 18, 67–74. [Google Scholar] [CrossRef]
- Hung, Y.H.; Chiu, W.C.; Fuh, S.R.; Lai, Y.T.; Tung, T.H.; Huang, C.C.; Lo, C.M. ECIS Based Electric Fence Method for Measurement of Human Keratinocyte Migration on Different Substrates. Biosensors 2022, 12, 293. [Google Scholar] [CrossRef]
- Ramasamy, S.; Bennet, D.; Kim, S. Drug and bioactive molecule screening based on a bioelectrical impedance cell culture platform. Int. J. Nanomed. 2014, 9, 5789–5809. [Google Scholar] [CrossRef]
- Sun, T.; Jackson, S.; Haycock, J.W.; Macneil, S. Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. J. Biotechnol. 2006, 122, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Bhadriraju, K.; Chen, C.S. Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov. Today 2002, 7, 612–620. [Google Scholar] [CrossRef]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G.; Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-Dimensional Cell Culture: A Breakthrough in Vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Laplante, A.F.; Germain, L.; Auger, F.A.; Moulin, V. Mechanisms of wound reepithelialization: Hints from a tissue-engineered reconstructed skin to long-standing questions. FASEB J. 2001, 15, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- El Ghalbzouri, A.; Hensbergen, P.; Gibbs, S.; Kempenaar, J.; van der Schors, R.; Ponec, M. Fibroblasts facilitate re-epithelialization in wounded human skin equivalents. Lab. Investig. 2004, 84, 102–112. [Google Scholar] [CrossRef]
- Breetveld, M.; Richters, C.D.; Rustemeyer, T.; Scheper, R.J.; Gibbs, S. Comparison of wound closure after burn and cold injury in human skin equivalents. J. Investig. Dermatol. 2006, 126, 1918–1921. [Google Scholar] [CrossRef]
- Xie, Y.; Rizzi, S.C.; Dawson, R.; Lynam, E.; Richards, S.; Leavesley, D.I.; Upton, Z. Development of a three-dimensional human skin equivalent wound model for investigating novel wound healing therapies. Tissue Eng. Part C. Methods 2010, 16, 1111–1123. [Google Scholar] [CrossRef]
- Egles, C.; Garlick, J.A.; Shamis, Y. Three-Dimensional Human Tissue Models of Wounded Skin. In Epidermal Cells: Methods in Molecular Biology; Turksen, K., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 585, pp. 345–359. ISBN 978-1-60761-379-4. [Google Scholar] [CrossRef]
- Blais, M.; Mottier, L.; Germain, M.-A.; Bellenfant, S.; Cadau, S.; Berthod, F. Sensory Neurons Accelerate Skin Reepithelialization via Substance P in an Innervated Tissue-Engineered Wound Healing Model. Tissue Eng. Part A 2014, 20, 2180–2188. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Iyer, K.; Chen, Z.; Ganapa, T.; Wu, B.M.; Tawil, B.; Linsley, C.S. Keratinocyte Migration in a Three-Dimensional In Vitro Wound Healing Model Co-Cultured with Fibroblasts. Tissue Eng. Regen. Med. 2018, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Graves Id, N.; Phillips Id, C.J.; Harding Id, K.; Graves, N. A narrative review of the epidemiology and economics of chronic wounds. Br. J. Dermatol. 2021, 187, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in chronic wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Nunan, R.; Harding, K.G.; Martin, P. Clinical challenges of chronic wounds: Searching for an optimal animal model to recapitulate their complexity. DMM Dis. Model. Mech. 2014, 7, 1205–1213. [Google Scholar] [CrossRef]
- Holzer-Geissler, J.C.J.; Schwingenschuh, S.; Zacharias, M.; Einsiedler, J.; Kainz, S.; Reisenegger, P.; Holecek, C.; Hofmann, E.; Wolff-Winiski, B.; Fahrngruber, H.; et al. Article The Impact of Prolonged Inflammation on Wound Healing. Biomedicines 2022, 10, 856. [Google Scholar] [CrossRef]
- Monika, P.; Chandraprabha, M.N.; Murthy, K.N.C.; Rangarajan, A.; Waiker, P.V.; Sathish, M. Human primary chronic wound derived fibroblasts demonstrate differential pattern in expression of fibroblast specific markers, cell cycle arrest and reduced proliferation. Exp. Mol. Pathol. 2022, 127, 104803. [Google Scholar] [CrossRef] [PubMed]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef]
- Schwarz, F.; Jennewein, M.; Bubel, M.; Holstein, J.H.; Pohlemann, T.; Oberringer, M. Soft tissue fibroblasts from well healing and chronic human wounds show different rates of myofibroblasts in vitro. Mol. Biol. Rep. 2013, 40, 1721–1733. [Google Scholar] [CrossRef]
- Caley, M.; Wall, I.B.; Peake, M.; Kipling, D.; Giles, P.; Thomas, D.W.; Stephens, P. Development and characterisation of a human chronic skin wound cell line—Towards an alternative for animal experimentation. Int. J. Mol. Sci. 2018, 19, 1001. [Google Scholar] [CrossRef]
- Maione, A.G.; Brudno, Y.; Stojadinovic, O.; Park, L.K.; Smith, A.; Tellechea, A.; Leal, E.C.; Kearney, C.J.; Veves, A.; Tomic-Canic, M.; et al. Three-Dimensional Human Tissue Models That Incorporate Diabetic Foot Ulcer-Derived Fibroblasts Mimic In Vivo Features of Chronic Wounds. Tissue Eng. Part C. Methods 2015, 21, 508–517. [Google Scholar] [CrossRef]
- Ozdogan, C.Y.; Kenar, H.; Davun, K.E.; Yucel, D.; Doger, E.; Alagoz, S. An in vitro 3D diabetic human skin model from diabetic primary cells. Biomed. Mater. 2021, 16, 015027. [Google Scholar] [CrossRef]
- Lim, I.J.; Phan, T.T.; Song, C.; Tan, W.T.L.; Longaker, M.T. Investigation of the influence of keloid-derived keratinocytes on fibroblast growth and proliferation in vitro. Plast. Reconstr. Surg. 2001, 107, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Funayama, E.; Chodon, T.; Oyama, A.; Sugihara, T. Keratinocytes Promote Proliferation and Inhibit Apoptosis of the Underlying Fibroblasts: An Important Role in the Pathogenesis of Keloid. J. Investig. Dermatol. 2003, 121, 1326–1331. [Google Scholar] [CrossRef]
- Butler, P.D.; Ly, D.P.; Longaker, M.T.; Yang, G.P. Use of organotypic coculture to study keloid biology. Am. J. Surg. 2008, 195, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Suttho, D.; Mankhetkorn, S.; Binda, D.; Pazart, L.; Humbert, P.; Rolin, G. 3D modeling of keloid scars in vitro by cell and tissue engineering. Arch. Dermatol. Res. 2017, 309, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Limandjaja, G.C.; van den Broek, L.J.; Breetveld, M.; Waaijman, T.; Monstrey, S.; de Boer, E.M.; Scheper, R.J.; Niessen, F.B.; Gibbs, S. Characterization of In Vitro Reconstructed Human Normotrophic, Hypertrophic, and Keloid Scar Models. Tissue Eng. Part C Methods 2018, 24, 242–253. [Google Scholar] [CrossRef]
- Limandjaja, G.C.; van den Broek, L.J.; Waaijman, T.; Breetveld, M.; Monstrey, S.; Scheper, R.J.; Niessen, F.B.; Gibbs, S. Reconstructed human keloid models show heterogeneity within keloid scars. Arch. Dermatol. Res. 2018, 310, 815–826. [Google Scholar] [CrossRef]
- Monsuur, H.N.; Boink, M.A.; Weijers, E.M.; Roffel, S.; Breetveld, M.; Gefen, A.; van den Broek, L.J.; Gibbs, S. Methods to study differences in cell mobility during skin wound healing in vitro. J. Biomech. 2016, 49, 1381–1387. [Google Scholar] [CrossRef]
- Limandjaja, G.C.; Waaijman, T.; Roffel, S.; Niessen, F.B.; Gibbs, S. Monocytes co-cultured with reconstructed keloid and normal skin models skew towards M2 macrophage phenotype. Arch. Dermatol. Res. 2019, 311, 615–627. [Google Scholar] [CrossRef]
- Phan, T.-T.; Sun, L.; Bay, B.-H.; Chan, S.-Y.; Lee, S.-T. Dietary Compounds Inhibit Proliferation and Contraction of Keloid and Hypertrophic Scar-Derived Fibroblasts In Vitro. J. Trauma Inj. Infect. Crit. Care 2003, 54, 1212–1224. [Google Scholar] [CrossRef]
- Wang, J.; Dodd, C.; Shankowsky, H.A.; Scott, P.G.; Tredget, E.E. Deep dermal fibroblasts contribute to hypertrophic scarring. Lab. Investig. 2008, 88, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Ciarmiello, L.F.; Mondola, P.; Damiano, S.; Seru, R.; Argenziano, C.; Nacca, M.; Santoriello, M.; Garbi, C. Differential p63 and p53 Expression in Human Keloid Fibroblasts and Hypertrophic Scar Fibroblasts. DNA Cell Biol. 2007, 26, 541–547. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Garbi, C.; Santoriello, M.; Santillo, A.; Wilson, R.R. Differential apoptosis markers in human keloids and hypertrophic scars fibroblasts. Mol. Cell. Biochem. 2009, 327, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Y.; Cheng, T.; Zheng, M.H.; Yi, C.G.; Pan, H.; Li, Z.J.; Chen, X.L.; Yu, Q.; Jiang, L.F.; Zhou, F.Y.; et al. Activation of peroxisome proliferator-activated receptor-inhibits transforming growth factor-1 induction of connective tissue growth factor and extracellular matrix in hypertrophic scar Wbroblasts in vitro. Arch Dermatol Res 2009, 301, 515–522. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.S.; Lee, J.W.; Choi, K.Y.; Yang, J.D.; Cho, B.C.; Oh, E.J.; Kim, T.J.; Ko, U.H.; Shin, J.H.; et al. Effect of Keratinocytes on Myofibroblasts in Hypertrophic Scars. Aesthetic Plast. Surg. 2019, 43, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Moulin, V.; Larochelle, S.; Langlois, C.; Thibault, I.; Lopez-Vallé, C.A.; Roy, M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J. Cell. Physiol. 2004, 198, 350–358. [Google Scholar] [CrossRef]
- Linge, C.; Richardson, J.; Vigor, C.; Clayton, E.; Hardas, B.; Rolfe, K.J. Hypertrophic scar cells fail to undergo a form of apoptosis specific to contractile collagen—The role of tissue transglutaminase. J. Investig. Dermatol. 2005, 125, 72–82. [Google Scholar] [CrossRef]
- Younai, S.; Nichter, L.S.; Wellisz, T.; Reinisch, J.; Nimni, M.E.; Tuan, T.L.; Tredget, E.E. Modulation of collagen synthesis by transforming growth factor-β in keloid and hypertrophic scar fibroblasts. Ann. Plast. Surg. 1994, 33, 148–154. [Google Scholar] [CrossRef]
- Simon, F.; Bergeron, D.; Larochelle, S.; Lopez-Vallé, C.A.; Genest, H.; Armour, A.; Moulin, V.J. Enhanced secretion of TIMP-1 by human hypertrophic scar keratinocytes could contribute to fibrosis. Burns 2012, 38, 421–427. [Google Scholar] [CrossRef]
- Bellemare, J.; Roberge, C.J.; Bergeron, D.; Lopez-Vallé, C.A.; Roy, M.; Moulin, V.J. Epidermis promotes dermal fibrosis: Role in the pathogenesis of hypertrophic scars. J. Pathol. 2005, 206, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lafosse, A.; Dufeys, C.; Beauloye, C.; Horman, S.; Dufrane, D. Impact of hyperglycemia and low oxygen tension on adipose-derived stem cells compared with dermal fibroblasts and keratinocytes: Importance for wound healing in type 2 diabetes. PLoS ONE 2016, 11, e0168058. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.C.E.; Liu, I.H.; Fang, A.H.; Wen, C.H.; Wu, C.S. Hyperglycaemic conditions decrease cultured keratinocyte mobility: Implications for impaired wound healing in patients with diabetes. Br. J. Dermatol. 2008, 159, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Guo, R.; Cheng, W.; Chai, L.; Wang, W.; Cao, C.; Li, S. High glucose inhibits ClC-2 chloride channels and attenuates cell migration of rat keratinocytes. Drug Des. Devel. Ther. 2015, 9, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.S.; Berends, R.F.; Flint, D.J.; Martin, P.E.M. Cell motility in models of wounded human skin is improved by Gap27 despite raised glucose, insulin and IGFBP-5. Exp. Cell Res. 2013, 319, 390–401. [Google Scholar] [CrossRef]
- Ueck, C.; Volksdorf, T.; Houdek, P.; Vidal-Y-Sy, S.; Sehner, S.; Ellinger, B.; Lobmann, R.; Larena-Avellaneda, A.; Reinshagen, K.; Ridderbusch, I.; et al. Comparison of in-vitro and ex-vivo wound healing assays for the investigation of diabetic wound healing and demonstration of a beneficial effect of a triterpene extract. PLoS ONE 2017, 12, e0169028. [Google Scholar] [CrossRef]
- Finnson, K.W.; McLean, S.; Di Guglielmo, G.M.; Philip, A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv. Wound Care 2013, 2, 195–214. [Google Scholar] [CrossRef]
- Mokos, Z.B.; Jović, A.; Grgurević, L.; Dumić-Čule, I.; Kostović, K.; Čeović, R.; Marinović, B. Current Therapeutic Approach to Hypertrophic Scars. Front. Med. 2017, 4, 83. [Google Scholar] [CrossRef]
- Robles, D.T.; Berg, D. Abnormal wound healing: Keloids. Clin. Dermatol. 2007, 25, 26–32. [Google Scholar] [CrossRef]
- Limandjaja, G.C.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. The Keloid Disorder: Heterogeneity, Histopathology, Mechanisms and Models. Front. Cell Dev. Biol. 2020, 8, 360. [Google Scholar] [CrossRef]
- Supp, D.M. Animal Models for Studies of Keloid Scarring. Adv. Wound Care 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, A.; Kamochi, H.; Sarukawa, S.; Uda, H.; Sugawara, Y.; Asahi, R.; Chi, D.; Nakagawa, S.; Kanayama, K.; Yoshimura, K. Reconstitution of Human Keloids in Mouse Skin. Plast. Reconstr. Surg. Glob. Open 2017, 5, e1304. [Google Scholar] [CrossRef] [PubMed]
- Polo, M.; Kim, Y.J.; Kucukcelebi, A.; Hayward, P.G.; Ko, F.; Robson, M.C. An in vivo model of human proliferative scar. J. Surg. Res. 1998, 74, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Shetlar, M.R.; Shetlar, C.L.; Kischer, C.W.; Pindur, J. Implants of keloid and hypertrophic scars into the athymic nude mouse: Changes in the glycosaminoglycans of the implants. Connect. Tissue Res. 1991, 26, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, S. Establishment of an animal model for human keloid scars using tissue engineering method. J. Burn Care Res. 2013, 34, 439–446. [Google Scholar] [CrossRef]
- Diegelmann, R.F.; Cohen, I.K.; McCoy, B.J. Growth kinetics and collagen synthesis of normal skin, normal scar and keloid fibroblasts in vitro. J. Cell. Physiol. 1979, 98, 341–346. [Google Scholar] [CrossRef]
- Cohen, I.K.; Diegelmann, R.F.; McCoy, B. Elevated collagen synthesis in cultured keloid fibroblasts. Surg. Forum 1977, 28, 526–527. [Google Scholar]
- Kischer, C.W.; Wagner, H.N.; Pindur, J.; Holubec, H.; Jones, M.; Ulreich, J.B.; Scuderi, P. Increased fibronectin production by cell lines from hypertrophic scar and keloid. Connect. Tissue Res. 1989, 23, 279–288. [Google Scholar] [CrossRef]
- Babu, M.; Diegelmann, R.; Oliver, N. Fibronectin is overproduced by keloid fibroblasts during abnormal wound healing. Mol. Cell. Biol. 1989, 9, 1642–1650. [Google Scholar] [CrossRef]
- Oliver, N.; Babu, M.; Diegelmann, R. Fibronectin gene transcription is enhanced in abnormal wound healing. J. Investig. Dermatol. 1992, 99, 572–578. [Google Scholar] [CrossRef]
- Meyer, L.J.M.; Egbert, B.M.; Shuster, S.; Stern, R.; Russell, S.B.; Russell, J.D.; Trupin, J.S. Reduced Hyaluronan in Keloid Tissue and Cultured Keloid Fibroblasts. J. Investig. Dermatol. 2003, 114, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Alaish, S.M.; Yager, D.R.; Diegelmann, R.F.; Cohen, I.K. Hyaluronic acid metabolism in keloid fibroblasts. J. Pediatr. Surg. 1995, 30, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Clore, J.N.; Cohen, I.K.; Diegelmann, R.F. Quantitative assay of types I and III collagen synthesized by keloid biopsies and fibroblasts. Biochim. Biophys. Acta—Gen. Subj. 1979, 586, 384–390. [Google Scholar] [CrossRef]
- Russell, J.D.; Witt, W.S. Cell size and growth characteristics of cultured fibroblasts isolated from normal and keloid tissue. Plast. Reconstr. Surg. 1976, 57, 207–212. [Google Scholar] [CrossRef]
- Oku, T.; Takigawa, M.; Yamada, M. Cell proliferation kinetics of cultured human keratinocytes and fibroblasts measured using a monoclonal antibody. Br. J. Dermatol. 1987, 116, 673–679. [Google Scholar] [CrossRef] [PubMed]
- MANCINI, R.E.; QUAIFE, J.V. Histogenesis of experimentally produced keloids. J. Investig. Dermatol. 1962, 38, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, L.Y.; Uitto, J.; Wortsman, J.; Abergel, R.P.; Dietrich, J. Ultrastructural Characteristics of Keloid Fibroblasts. Am. J. Dermatopathol. 1988, 10, 505–508. [Google Scholar] [CrossRef]
- Calderon, M.; Lawrence, W.T.; Banes, A.J. Increased proliferation in keloid fibroblasts wounded in vitro. J. Surg. Res. 1996, 61, 343–347. [Google Scholar] [CrossRef]
- Maas-Szabowski, N.; Stark, H.-J.; Fusenig, N.E. Keratinocyte Growth Regulation in Defined Organotypic Cultures Through IL-1-Induced Keratinocyte Growth Factor Expression in Resting Fibroblasts. J. Investig. Dermatol. 2000, 114, 1075–1084. [Google Scholar] [CrossRef]
- Maas-Szabowski, N.; Shimotoyodome, A.; Fusenig, N.E. Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. J. Cell Sci. 1999, 112 Pt 12, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Szabowski, A.; Maas-Szabowski, N.; Andrecht, S.; Kolbus, A.; Schorpp-Kistner, M.; Fusenig, N.E.; Angel, P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell 2000, 103, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Garner, W.L. Epidermal Regulation of Dermal Fibroblast Activity. Plast. Reconstr. Surg. 1998, 102, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Lim, I.J.; Phan, T.T.; Bay, B.H.; Qi, R.; Huynh, H.-T.; Tan, W.T.L.; Lee, S.T.; Longaker, M.T. Fibroblasts cocultured with keloid keratinocytes: Normal fibroblasts secrete collagen in a keloidlike manner. Am. J. Physiol.—Cell Physiol. 2002, 283, C212–C222. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.-T.; Lim, I.J.; Bay, B.-H.; Qi, R.; Huynh, H.T.; Lee, S.-T.; Longaker, M.T. Differences in collagen production between normal and keloid-derived fibroblasts in serum-media co-culture with keloid-derived keratinocytes. J. Dermatol. Sci. 2002, 29, 26–34. [Google Scholar] [CrossRef]
- Hahn, J.M.; Glaser, K.; McFarland, K.L.; Aronow, B.J.; Boyce, S.T.; Supp, D.M. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013, 21, 530–544. [Google Scholar] [CrossRef]
- Rosen, D.J.; Patel, M.K.; Freeman, K.; Weiss, P.R. A primary protocol for the management of ear keloids: Results of excision combined with intraoperative and postoperative steroid injections. Plast. Reconstr. Surg. 2007, 120, 1395–1400. [Google Scholar] [CrossRef]
- Golladay, E.S. Treatment of keloids by single intraoperative perilesional injection of repository steroid. South. Med. J. 1988, 81, 736–738. [Google Scholar] [CrossRef]
- Hietanen, K.E.; Järvinen, T.A.; Huhtala, H.; Tolonen, T.T.; Kuokkanen, H.O.; Kaartinen, I.S. Treatment of keloid scars with intralesional triamcinolone and 5-fluorouracil injections—A randomized controlled trial. J. Plast. Reconstr. Aesthetic Surg. 2019, 72, 4–11. [Google Scholar] [CrossRef]
- Darougheh, A.; Asilian, A.; Shariati, F. Intralesional triamcinolone alone or in combination with 5-fluorouracil for the treatment of keloid and hypertrophic scars. Clin. Exp. Dermatol. 2009, 34, 219–223. [Google Scholar] [CrossRef]
- Apikian, M.; Goodman, G. Intralesional 5-fluorouracil in the treatment of keloid scars. Australas. J. Dermatol. 2004, 45, 140–143. [Google Scholar] [CrossRef]
- Khalid, F.A.; Mehrose, M.Y.; Saleem, M.; Yousaf, M.A.; Mujahid, A.M.; Rehman, S.U.; Ahmad, S.; Tarar, M.N. Comparison of efficacy and safety of intralesional triamcinolone and combination of triamcinolone with 5-fluorouracil in the treatment of keloids and hypertrophic scars: Randomised control trial. Burns 2019, 45, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Bayat, A. Superior effect of combination vs. single steroid therapy in keloid disease: A comparative in vitro analysis of glucocorticoids. Wound Repair Regen. 2013, 21, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Cai, Y.J.; Lung, I.; Leung, B.C.S.; Burd, A. A study of the combination of triamcinolone and 5-fluorouracil in modulating keloid fibroblasts in vitro. J. Plast. Reconstr. Aesthetic Surg. 2013, 66, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kalra, A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology 2002, 204, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, R.E. Treatment of inflamed hypertrophic scars using intralesional 5-FU. Dermatol. Surg. 1999, 25, 224–232. [Google Scholar] [CrossRef]
- Son, Y.; Phillips, E.O.N.; Price, K.M.; Rosenberg, L.Z.; Stefanovic, B.; Wolfe, C.M.; Shaath, T.S.; Om, A.; Cohen, G.F.; Gunjan, A. Treatment of keloids with a single dose of low-energy superficial X-ray radiation to prevent recurrence after surgical excision: An in vitro and in vivo study. J. Am. Acad. Dermatol. 2020, 83, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Rössler, S.; Nischwitz, S.P.; Luze, H.; Holzer-Geissler, J.C.J.; Zrim, R.; Kamolz, L.P. In Vivo Models for Hypertrophic Scars—A Systematic Review. Medicina 2022, 58, 736. [Google Scholar] [CrossRef]
- van den Broek, L.J.; Limandjaja, G.C.; Niessen, F.B.; Gibbs, S. Human hypertrophic and keloid scar models: Principles, limitations and future challenges from a tissue engineering perspective. Exp. Dermatol. 2014, 23, 382–386. [Google Scholar] [CrossRef]
- Ramos, M.L.C.; Gragnani, A.; Ferreira, L.M. Is there an ideal animal model to study hypertrophic scarring? J. Burn Care Res. 2008, 29, 363–368. [Google Scholar] [CrossRef]
- Zhu, K.Q.; Engrav, L.H.; Tamura, R.N.; Cole, J.A.; Muangman, P.; Carrougher, G.J.; Gibran, N.S. Further similarities between cutaneous scarring in the female, red Duroc pig and human hypertrophic scarring. Burns 2004, 30, 518–530. [Google Scholar] [CrossRef]
- Blackstone, B.N.; Kim, J.Y.; McFarland, K.L.; Sen, C.K.; Supp, D.M.; Bailey, J.K.; Powell, H.M. Scar formation following excisional and burn injuries in a red Duroc pig model. Wound Repair Regen. 2017, 25, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Gallant-Behm, C.L.; Hart, D.A. Genetic analysis of skin wound healing and scarring in a porcine model. Wound Repair Regen. 2006, 14, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Nischwitz, S.P.; Fink, J.; Schellnegger, M.; Luze, H.; Bubalo, V.; Tetyczka, C.; Roblegg, E.; Holecek, C.; Zacharias, M.; Kamolz, L.; et al. The Role of Local Inflammation and Hypoxia in the Formation of Hypertrophic Scars—A New Model in the Duroc Pig. Int. J. Mol. Sci. 2023, 24, 316. [Google Scholar] [CrossRef] [PubMed]
- Honardoust, D.; Kwan, P.; Momtazi, M.; Ding, J.; Tredget, E.E. Novel methods for the investigation of human hypertrophic scarring and other dermal fibrosis. Methods Mol. Biol. 2013, 1037, 203–231. [Google Scholar] [CrossRef]
- Varkey, M.; Ding, J.; Tredget, E.E. Differential collagen-glycosaminoglycan matrix remodeling by superficial and deep dermal fibroblasts: Potential therapeutic targets for hypertrophic scar. Biomaterials 2011, 32, 7581–7591. [Google Scholar] [CrossRef]
- Derderian, C.A.; Bastidas, N.; Lerman, O.Z.; Bhatt, K.A.; Lin, S.E.; Voss, J.; Holmes, J.W.; Levine, J.P.; Gurtner, G.C. Mechanical strain alters gene expression in an in vitro model of hypertrophic scarring. Ann. Plast. Surg. 2005, 55, 69–75. [Google Scholar] [CrossRef]
- Van Den Broek, L.J.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. Development, validation, and testing of a human tissue engineered hypertrophic scar model. ALTEX 2012, 29, 389–402. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Wang, Z.; Xia, Y.; Zhou, M.; Zhong, A.; Sun, J. Experimental models for cutaneous hypertrophic scar research. Wound Repair Regen. 2020, 28, 126–144. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef]
- Trottier, V.; Marceau-Fortier, G.; Germain, L.; Vincent, C.; Fradette, J. IFATS Collection: Using Human Adipose-Derived Stem/Stromal Cells for the Production of New Skin Substitutes. Stem Cells 2008, 26, 2713–2723. [Google Scholar] [CrossRef]
- Bellas, E.; Seiberg, M.; Garlick, J.; Kaplan, D.L. In vitro 3D Full-Thickness Skin-Equivalent Tissue Model Using Silk and Collagen Biomaterials. Macromol. Biosci. 2012, 12, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Monfort, A.; Soriano-Navarro, M.; García-Verdugo, J.M.; Izeta, A. Production of human tissue-engineered skin trilayer on a plasma-based hypodermis. J. Tissue Eng. Regen. Med. 2013, 7, 479–490. [Google Scholar] [CrossRef]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-derived stem cells for regenerative medicine. Circ. Res. 2007, 100, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Satija, N.K.; Gurudutta, G.U.; Sharma, S.; Afrin, F.; Gupta, P.; Verma, Y.K.; Singh, V.K.; Tripathi, R.P. Mesenchymal stem cells: Molecular targets for tissue engineering. Stem Cells Dev. 2007, 16, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Huber, B.; Link, A.; Linke, K.; Gehrke, S.A.; Winnefeld, M.; Kluger, P.J. Integration of Mature Adipocytes to Build-Up a Functional Three-Layered Full-Skin Equivalent. Tissue Eng.—Part C Methods 2016, 22, 756–764. [Google Scholar] [CrossRef]
- Huber, B.; Borchers, K.; Tovar, G.E.M.; Kluger, P.J. Methacrylated gelatin and mature adipocytes are promising components for adipose tissue engineering. J. Biomater. Appl. 2015, 30, 699–710. [Google Scholar] [CrossRef]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef]
- Vidal, S.E.L.; Tamamoto, K.A.; Nguyen, H.; Abbott, R.D.; Cairns, D.M.; Kaplan, D.L. 3D biomaterial matrix to support long term, full thickness, immuno-competent human skin equivalents with nervous system components. Biomaterials 2019, 198, 194–203. [Google Scholar] [CrossRef]
- Groeber, F.; Engelhardt, L.; Lange, J.; Kurdyn, S.; Schmid, F.F.; Rücker, C.; Mielke, S.; Walles, H.; Hansmann, J. A first vascularized skin equivalent for as an alternative to animal experimentation. ALTEX 2016, 33, 415–422. [Google Scholar] [CrossRef]
- Kim, B.S.; Gao, G.; Kim, J.Y.; Cho, D. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Adv. Healthc. Mater. 2019, 8, 1801019. [Google Scholar] [CrossRef]
- Matai, I.; Kaur, G.; Seyedsalehi, A.; McClinton, A.; Laurencin, C.T. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials 2020, 226, 119536. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Abaci, H.E.; Guo, Z.; Coffman, A.; Gillette, B.; Lee, W.H.; Sia, S.K.; Christiano, A.M. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv. Healthc. Mater. 2016, 5, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Pupovac, A.; Senturk, B.; Griffoni, C.; Maniura-Weber, K.; Rottmar, M.; McArthur, S.L. Toward Immunocompetent 3D Skin Models. Adv. Healthc. Mater. 2018, 7, 1701405. [Google Scholar] [CrossRef]
- Ouwehand, K.; Spiekstra, S.W.; Waaijman, T.; Scheper, R.J.; de Gruijl, T.D.; Gibbs, S. Technical Advance: Langerhans cells derived from a human cell line in a full-thickness skin equivalent undergo allergen-induced maturation and migration. J. Leukoc. Biol. 2011, 90, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Kühbacher, A.; Henkel, H.; Stevens, P.; Grumaz, C.; Finkelmeier, D.; Burger-Kentischer, A.; Sohn, K.; Rupp, S. Central Role for Dermal Fibroblasts in Skin Model Protection against Candida albicans. J. Infect. Dis. 2017, 215, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.Y.S.; Johnson, C.; Macneil, S.; Haycock, J.W.; Ghaemmaghami, A.M. The development of a 3D immunocompetent model of human skin. Biofabrication 2013, 5, 035011. [Google Scholar] [CrossRef]
- Bechetoille, N.; Dezutter-Dambuyant, C.; Damour, O.; André, V.; Orly, I.; Perrier, E. Effects of Solar Ultraviolet Radiation on Engineered Human Skin Equivalent Containing Both Langerhans Cells and Dermal Dendritic Cells. Tissue Eng. 2007, 13, 2667–2679. [Google Scholar] [CrossRef]
- Schellenberger, M.T.; Bock, U.; Hennen, J.; Groeber-Becker, F.; Walles, H.; Blömeke, B. A coculture system composed of THP-1 cells and 3D reconstructed human epidermis to assess activation of dendritic cells by sensitizing chemicals after topical exposure. Toxicol. Vitr. 2019, 57, 62–66. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Higgins, C.A.; Gillette, B.M.; Itoh, M.; Umegaki, N.; Gledhill, K.; Sia, S.K.; Christiano, A.M. Building a microphysiological skin model from induced pluripotent stem cells. Stem Cell Res. Ther. 2013, 4, S2. [Google Scholar] [CrossRef] [PubMed]
- Kashpur, O.; Smith, A.; Gerami-Naini, B.; Maione, A.G.; Calabrese, R.; Tellechea, A.; Theocharidis, G.; Liang, L.; Pastar, I.; Tomic-Canic, M.; et al. Differentiation of diabetic foot ulcer–derived induced pluripotent stem cells reveals distinct cellular and tissue phenotypes. FASEB J. 2019, 33, 1262. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.L.; Wang, S.; Yeong, W.Y.; Naing, M.W. Skin Bioprinting: Impending Reality or Fantasy? Trends Biotechnol. 2016, 34, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.; Metral, E.; Boher, A.; Rousselle, P.; Thepot, A.; Damour, O. In vitro 3-D model based on extending time of culture for studying chronological epidermis aging. Matrix Biol. 2015, 47, 85–97. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| In Vitro Model | Cells | Medium/Matrix | Limitations | Ref. | |
|---|---|---|---|---|---|
| Chronic wounds | 2D | Chronic-wound-derived fibroblasts (venous leg ulcer; undefined) | - | Lack of cell–cell interaction/vasculature/cell-to-environment interface; not immunocompetent | [59,60,61] |
| hTERT chronic wound fibroblast cell line (venous leg ulcer) | - | Lack of cell–cell interaction/vasculature/cell-to-environment interface; not immunocompetent | [62] | ||
| 3D | Fibroblasts from diabetic foot ulcers; NKs; endothelial cells | Collagen type I | Lack of vasculature; not immunocompetent | [63] | |
| Patient-derived (type 2 diabetes) dermal NFs; NKs; HUVECs | Hydrogel | Not immunocompetent | [64] | ||
| Keloids | 2D | Co-culturing NFs and KFs; NKs | - | Lack of vasculature/cell-to-environment interface; not immunocompetent | [65,66] |
| 3D | NFs and KFs; NFs | Collagen | Lack of vasculature; not immunocompetent | [67] | |
| KFs of different origins | Collagen | Lack of vasculature; not immunocompetent | [68] | ||
| Donor-matched KFs and KKs | Collagen-elastin | Lack of vasculature; not immunocompetent | [69,70,71] | ||
| KKs and KFs; CD14+ monocytes | Collagen-elastin | Lack of vasculature | [72] | ||
| Hypertrophic scars | 2D | HSFs | - | Lack of vasculature/cell-to-environment interface; not immunocompetent | [73,74,75,76,77] |
| Hmyo and NKs | - | Lack of vasculature/cell-to-environment interface; not immunocompetent | [78,79] | ||
| 3D | HSFs | Collagen | Lack of vasculature; not immunocompetent | [73,80] | |
| HSFs | Fibrin | Lack of vasculature; not immunocompetent | [81] | ||
| Hmyo, NFs, Wmyo; NKs | Manipulable sheets | Lack of vasculature; not immunocompetent | [82] | ||
| Wmyo, Hmyo, NFs; NKs or HSKs | Manipulable sheets | Lack of vasculature; not immunocompetent | [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofmann, E.; Fink, J.; Pignet, A.-L.; Schwarz, A.; Schellnegger, M.; Nischwitz, S.P.; Holzer-Geissler, J.C.J.; Kamolz, L.-P.; Kotzbeck, P. Human In Vitro Skin Models for Wound Healing and Wound Healing Disorders. Biomedicines 2023, 11, 1056. https://doi.org/10.3390/biomedicines11041056
Hofmann E, Fink J, Pignet A-L, Schwarz A, Schellnegger M, Nischwitz SP, Holzer-Geissler JCJ, Kamolz L-P, Kotzbeck P. Human In Vitro Skin Models for Wound Healing and Wound Healing Disorders. Biomedicines. 2023; 11(4):1056. https://doi.org/10.3390/biomedicines11041056
Chicago/Turabian StyleHofmann, Elisabeth, Julia Fink, Anna-Lisa Pignet, Anna Schwarz, Marlies Schellnegger, Sebastian P. Nischwitz, Judith C. J. Holzer-Geissler, Lars-Peter Kamolz, and Petra Kotzbeck. 2023. "Human In Vitro Skin Models for Wound Healing and Wound Healing Disorders" Biomedicines 11, no. 4: 1056. https://doi.org/10.3390/biomedicines11041056
APA StyleHofmann, E., Fink, J., Pignet, A.-L., Schwarz, A., Schellnegger, M., Nischwitz, S. P., Holzer-Geissler, J. C. J., Kamolz, L.-P., & Kotzbeck, P. (2023). Human In Vitro Skin Models for Wound Healing and Wound Healing Disorders. Biomedicines, 11(4), 1056. https://doi.org/10.3390/biomedicines11041056