
Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype

 , , , , , ,
, , , , , ,
Abstract
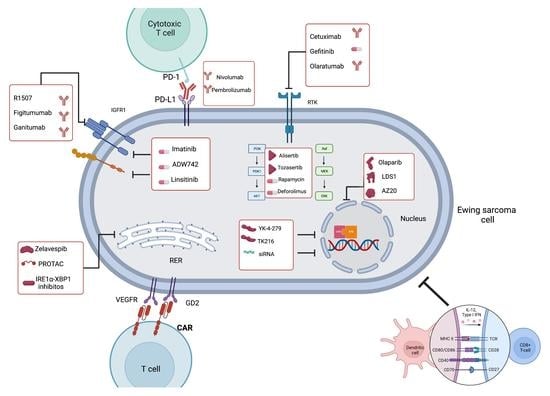
1. Introduction
2. Receptor Tyrosine Kinases (RTKs)
2.1. Insulin-like Growth Factor 1 Receptor (IGF1R) in ES
- Monoclonal antibodies directed against IGF1R
- Tyrosine kinase inhibitors against IGF1R
2.2. Other Targetable RTKs in ES
3. EWSR1-FLI1
3.1. Decreasing EWSR1-FLI1 Expression
3.2. Decreasing the Activity of EWSR1-FLI1
4. The Epigenomic Landscape of ES
4.1. Histones Demethylation Inhibitors (HDMi)
4.2. Histones Methyltransferase Inhibitors (HMTi)
4.3. Histones Deacetylase Inhibition (HDACi)
5. DNA Damage Response in ES
Poly (ADP-Ribose) Polymerase (PARP) Inhibitors (PARPi) in ES
6. The Downstream Effectors of EWSR1-FLI1
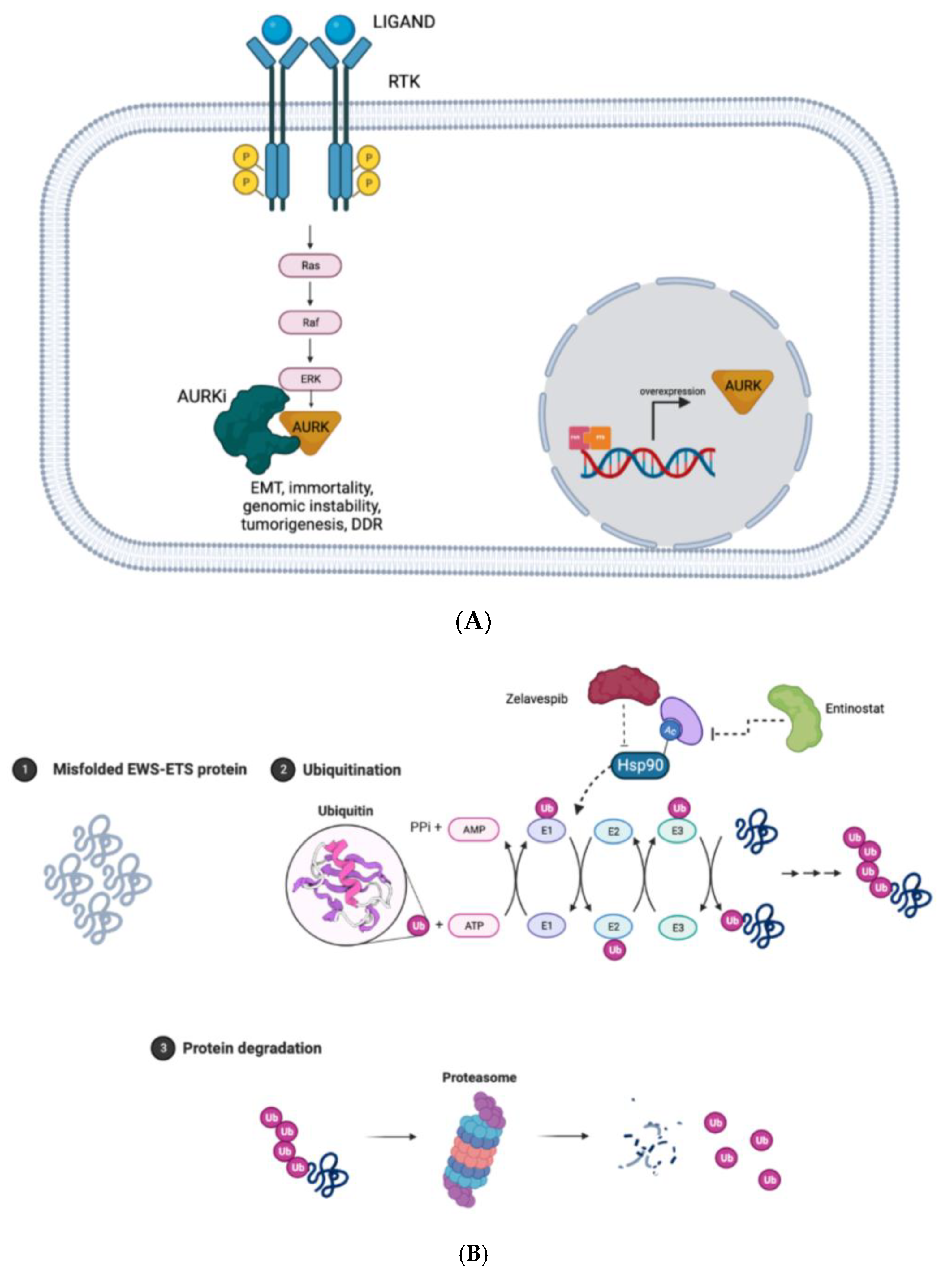
6.1. Aurora Kinase (AURK) Inhibitors
6.2. Forkhead Box O (FOXO)1
6.3. Glioma-Associated Oncogene Homolog 1 (Gli1)
6.4. Mammalian Target of Rapamycin (mTOR)
7. Homeostasis and Metabolism in ES
7.1. Heat-Shock Protein 90 (HSP90)
7.2. Proteolysis Targeting Chimeric Molecules (PROTACs)
7.3. Unfolded Protein Response and IRE1α-XBP1 Inhibitors
7.4. Warburg Effect Inhibition: Lactate Dehydrogenase Inhibitors (LDHi)
8. Immunotherapy in ES
8.1. Immune Checkpoint Inhibitors (ICIs)
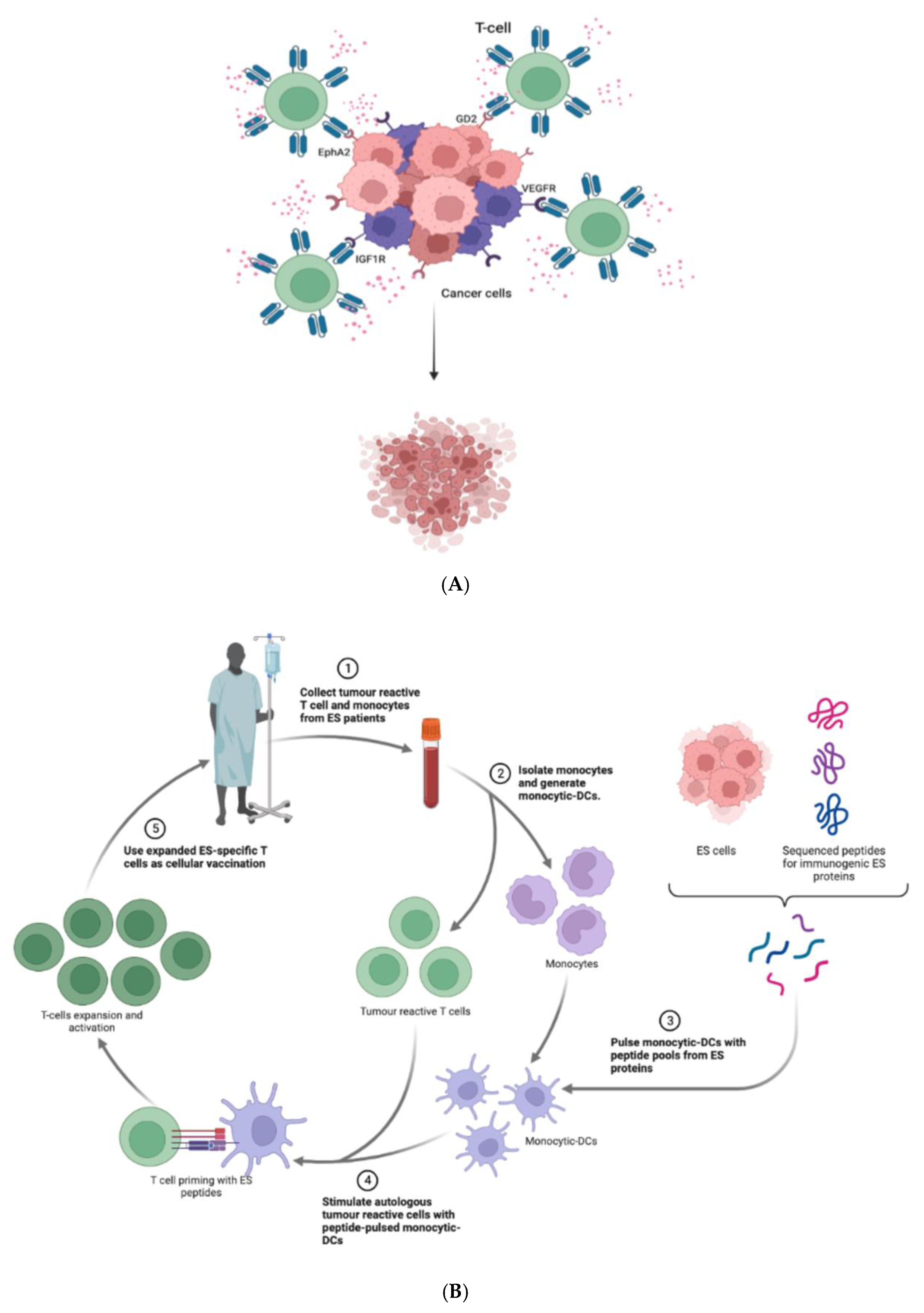
8.2. Tumor-Reactive T-Cells
8.3. Chimeric Antigen Receptor T-Cell (CAR-T)
8.4. Cancer Vaccines
9. Discussion
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ludwig, J.A. Ewing sarcoma: Historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr. Opin. Oncol. 2008, 20, 412–418. [Google Scholar] [CrossRef]
- Covello, B.; Hartman, S.; Kaufman, S.; Enrizo, O. Radiological and pathological diagnosis of an incidental Askin tumor. Radiol. Case Rep. 2021, 16, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Vural, Ç.; Uluoğlu, Ö.; Akyürek, N.; Oğuz, A.; Karadeniz, C. The Evaluation of CD99 Immunoreactivity and EWS/FLI1 Translocation by Fluorescence in situ Hybridization in Central PNETs and Ewing’s Sarcoma Family of Tumors. Pathol. Oncol. Res. 2011, 17, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.; Lessnick, S.L. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2012, 32, 5089–5100. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Stamenkovic, I. The Biology of Ewing sarcoma. Cancer Lett. 2007, 254, 1–10. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Schuck, A.; Ahrens, S.; Paulussen, M.; Kuhlen, M.; Könemann, S.; Rübe, C.; Winkelmann, W.; Kotz, R.; Dunst, J.; Willich, N.; et al. Local therapy in localized Ewing tumors: Results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int. J. Radiat. Oncol. 2003, 55, 168–177. [Google Scholar] [CrossRef]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.S.; Pawel, B.R.; Grier, H.E.; Marcus, K.; Sailer, S.; Healey, J.H.; Dormans, J.P.; et al. Randomized Controlled Trial of Interval-Compressed Chemotherapy for the Treatment of Localized Ewing Sarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef]
- Le Deley, M.-C.; Paulussen, M.; Lewis, I.; Brennan, B.; Ranft, A.; Whelan, J.; Le Teuff, G.; Michon, J.; Ladenstein, R.; Marec-Bérard, P.; et al. Cyclophosphamide Compared With Ifosfamide in Consolidation Treatment of Standard-Risk Ewing Sarcoma: Results of the Randomized Noninferiority Euro-EWING99-R1 Trial. J. Clin. Oncol. 2014, 32, 2440–2448. [Google Scholar] [CrossRef]
- McCabe, M.; Kirton, L.; Khan, M.; Fenwick, N.; Strauss, S.J.; Valverde, C.; Mata, C.; Gaspar, N.; Luksch, R.; Longhi, A.; et al. Phase III assessment of topotecan and cyclophosphamide and high-dose ifosfamide in rEECur: An international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J. Clin. Oncol. 2022, 40, LBA2. [Google Scholar] [CrossRef]
- Van Der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor Protein-Tyrosine Kinases and Their Signal Transduction Pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Yee, D.; Favoni, R.E.; Lebovic, G.S.; Lombana, F.; Powell, D.R.; Reynolds, C.P.; Rosen, N. Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation. A potential autocrine growth factor. J. Clin. Investig. 1990, 86, 1806–1814. [Google Scholar] [CrossRef]
- Cironi, L.; Riggi, N.; Provero, P.; Wolf, N.; Suvà, M.-L.; Suvà, D.; Kindler, V.; Stamenkovic, I. IGF1 Is a Common Target Gene of Ewing’s Sarcoma Fusion Proteins in Mesenchymal Progenitor Cells. PLoS ONE 2008, 3, e2634. [Google Scholar] [CrossRef]
- France, K.A.; Anderson, J.L.; Park, A.; Denny, C.T. Oncogenic Fusion Protein EWS/FLI1 Down-regulates Gene Expression by Both Transcriptional and Posttranscriptional Mechanisms. J. Biol. Chem. 2011, 286, 22750–22757. [Google Scholar] [CrossRef]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 Silencing and Gene Profiling of Ewing Cells Reveal Downstream Oncogenic Pathways and a Crucial Role for Repression of Insulin-Like Growth Factor Binding Protein 3. Mol. Cell. Biol. 2004, 24, 7275–7283. [Google Scholar] [CrossRef] [PubMed]
- de Alava, E.; Panizo, A.; Antonescu, C.R.; Huvos, A.G.; Pardo-Mindán, F.J.; Barr, F.G.; Ladanyi, M. Association of EWS-FLI1 Type 1 Fusion with Lower Proliferative Rate in Ewing’s Sarcoma. Am. J. Pathol. 2000, 156, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.-P.; Chawla, S.P.; Toner, G.; Maki, R.G.; Meyers, P.; Chugh, R.; et al. R1507, a Monoclonal Antibody to the Insulin-Like Growth Factor 1 Receptor, in Patients With Recurrent or Refractory Ewing Sarcoma Family of Tumors: Results of a Phase II Sarcoma Alliance for Research Through Collaboration Study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Houghton, P.J.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wu, J.; Smith, M.A. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 54, 921–926. [Google Scholar] [CrossRef]
- Scartozzi, M.; Bianconi, M.; Maccaroni, E.; Giampieri, R.; Berardi, R.; Cascinu, S. Dalotuzumab, a recombinant humanized mAb targeted against IGFR1 for the treatment of cancer. Curr. Opin. Mol. Ther. 2010, 12, 361–371. [Google Scholar] [PubMed]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II Study of Ganitumab, a Fully Human Anti–Type-1 Insulin-Like Growth Factor Receptor Antibody, in Patients With Metastatic Ewing Family Tumors or Desmoplastic Small Round Cell Tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef]
- DuBois, S.G.; Krailo, M.D.; Glade-Bender, J.; Buxton, A.; Laack, N.; Randall, R.L.; Chen, H.X.; Seibel, N.L.; Boron, M.; Terezakis, S.; et al. Randomized Phase III Trial of Ganitumab With Interval-Compressed Chemotherapy for Patients With Newly Diagnosed Metastatic Ewing Sarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2023. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary Efficacy of the Anti-Insulin–Like Growth Factor Type 1 Receptor Antibody Figitumumab in Patients With Refractory Ewing Sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.G.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Pápai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Gomez, N.C.; McFadden, A.W.; Moats-Staats, B.M.; Wu, S.; Rojas, A.; Sapp, T.; Simon, J.M.; Smith, S.V.; Kaiser-Rogers, K.; et al. PTEN deficiency mediates a reciprocal response to IGFI and mTOR inhibition. Mol. Cancer Res. 2014, 12, 1610–1620. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., II; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.S.; Mackintosh, C.; Martin, D.H.; Campos, M.; Hernández, T.; Ordóñez, J.-L.; de Alava, E. Insulin-Like Growth Factor I Receptor Pathway Inhibition by ADW742, Alone or in Combination with Imatinib, Doxorubicin, or Vincristine, Is a Novel Therapeutic Approach in Ewing Tumor. Clin. Cancer Res. 2006, 12, 3532–3540. [Google Scholar] [CrossRef]
- Manara, M.C.; Perdichizzi, S.; Serra, M.; Pierini, R.; Benini, S.; Hattinger, C.M.; Astolfi, A.; Bagnati, R.; D’Incalci, M.; Picci, P.; et al. The molecular mechanisms responsible for resistance to ET-743 (Trabectidin; Yondelis) in the Ewing’s sarcoma cell line, TC-71. Int. J. Oncol. 2005, 27, 1605–1616. [Google Scholar]
- Benini, S.; Manara, M.C.; Baldini, N.; Cerisano, V.; Serra, M.; Mercuri, M.; Lollini, P.L.; Nanni, P.; Picci, P.; Scotlandi, K. Inhibition of insulin-like growth factor I receptor increases the antitumor activity of doxorubicin and vincristine against Ewing’s sarcoma cells. Clin. Cancer Res. 2001, 7, 1790–1797. [Google Scholar]
- Jones, R.L.; Kim, E.S.; Nava-Parada, P.; Alam, S.; Johnson, F.M.; Stephens, A.W.; Simantov, R.; Poondru, S.; Gedrich, R.; Lippman, S.M.; et al. Phase I Study of Intermittent Oral Dosing of the Insulin-like Growth Factor-1 and Insulin Receptors Inhibitor OSI-906 in Patients With Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 693–700. [Google Scholar] [CrossRef]
- Amaral, A.T.; Garofalo, C.; Frapolli, R.; Manara, M.C.; Mancarella, C.; Uboldi, S.; Di Giandomenico, S.; Ordóñez, J.L.; Sevillano, V.; Malaguarnera, R.; et al. Trabectedin Efficacy in Ewing Sarcoma Is Greatly Increased by Combination with Anti-IGF Signaling Agents. Clin. Cancer Res. 2015, 21, 1373–1382. [Google Scholar] [CrossRef]
- Scotlandi, K.; Manara, M.C.; Nicoletti, G.; Lollini, P.-L.; Lukas, S.; Benini, S.; Croci, S.; Perdichizzi, S.; Zambelli, D.; Serra, M.; et al. Antitumor Activity of the Insulin-Like Growth Factor-I Receptor Kinase Inhibitor NVP-AEW541 in Musculoskeletal Tumors. Cancer Res. 2005, 65, 3868–3876. [Google Scholar] [CrossRef]
- del Rincon, J.-P.; Iida, K.; Gaylinn, B.D.; McCurdy, C.E.; Leitner, J.W.; Barbour, L.A.; Kopchick, J.J.; Friedman, J.E.; Draznin, B.; Thorner, M.O. Growth Hormone Regulation of p85α Expression and Phosphoinositide 3-Kinase Activity in Adipose Tissue. Diabetes 2007, 56, 1638–1646. [Google Scholar] [CrossRef]
- Chao, J.; Budd, G.T.; Chu, P.; Frankel, P.; Garcia, D.; Junqueira, M.; Loera, S.; Somlo, G.; Sato, J.; Chow, W.A. Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer Res. 2010, 30, 547–552. [Google Scholar]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Knoche, S.; Stephens, J.; Li, H.; Surguladze, D.; Loizos, N.; Luffer-Atlas, D.; Oakley, G.J.; et al. Olaratumab Exerts Antitumor Activity in Preclinical Models of Pediatric Bone and Soft Tissue Tumors through Inhibition of Platelet-Derived Growth Factor Receptor α. Clin. Cancer Res. 2018, 24, 847–857. [Google Scholar] [CrossRef]
- Ahmed, A.; Gilbert-Barness, E.; Lacson, A. Expression of C-kit in Ewing Family of Tumors: A Comparison of Different Immunohistochemical Protocols. Pediatr. Dev. Pathol. 2004, 7, 342–347. [Google Scholar] [CrossRef]
- Daw, N.C.; Furman, W.L.; Stewart, C.F.; Iacono, L.C.; Krailo, M.; Bernstein, M.L.; Dancey, J.E.; Speights, R.A.; Blaney, S.M.; Croop, J.M.; et al. Phase I and Pharmacokinetic Study of Gefitinib in Children With Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2005, 23, 6172–6180. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, H.; Wu, C.; Jiang, Y.; Xiao, L.; Tian, Y. Overcoming cetuximab resistance in Ewing’s sarcoma by inhibiting lactate dehydrogenase-A. Mol. Med. Rep. 2016, 14, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xie, L.; Sun, X.; Liu, K.; Tang, X.; Yan, T.; Yang, R.; Guo, W.; Gu, J. Anlotinib, Vincristine, and Irinotecan for Advanced Ewing Sarcoma After Failure of Standard Multimodal Therapy: A Two-Cohort, Phase Ib/II Trial. Oncologist 2021, 26, e1256–e1262. [Google Scholar] [CrossRef] [PubMed]
- van der Ent, W.; Sand, L.G.; Hogendoorn, P.C. Molecular genetics of Ewing sarcoma, model systems and finding novel (immuno-) therapeutic targets. J. Transl. Genet. Genom. 2018, 2, 10. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Subhan, A.; Torchilin, V. siRNA based drug design, quality, delivery and clinical translation. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102239. [Google Scholar] [CrossRef] [PubMed]
- Simmons, O.; Maples, P.B.; Senzer, N.; Nemunaitis, J. Ewing’s Sarcoma: Development of RNA Interference-Based Therapy for Advanced Disease. ISRN Oncol. 2012, 2012, 247657. [Google Scholar] [CrossRef]
- Rao, D.D.; Jay, C.; Wang, Z.; Luo, X.; Kumar, P.; Eysenbach, H.; Ghisoli, M.; Senzer, N.; Nemunaitis, J. Preclinical Justification of pbi-shRNA EWS/FLI1 Lipoplex (LPX) Treatment for Ewing’s Sarcoma. Mol. Ther. 2016, 24, 1412–1422. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Erkizan, V.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Üren, A. Oncoprotein EWS-FLI1 Activity Is Enhanced by RNA Helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.-E.; Menegaz, B.A.; Ramamoorthy, V.; Aiyer, R.A.; Maywald, R.L.; Buford, A.S.; Doolittle, D.K.; Culotta, K.S.; O’Dorisio, J.E.; Ludwig, J.A. An Oral Formulation of YK-4-279: Preclinical Efficacy and Acquired Resistance Patterns in Ewing Sarcoma. Mol. Cancer Ther. 2015, 14, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.; Federman, N.C.; Anderson, P.M.; Macy, M.E.; Riedel, R.F.; Davis, L.E.; Daw, N.C.; Wulff, J.; Kim, A.; Ratan, R.; et al. TK216 for relapsed/refractory Ewing sarcoma: Interim phase 1/2 results. J. Clin. Oncol. 2021, 39, 11500. [Google Scholar] [CrossRef]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 Utilizes Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar] [CrossRef]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 Inhibition Interferes with Global EWS/ETS Transcriptional Activity and Impedes Ewing Sarcoma Tumor Growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef]
- Schildhaus, H.-U.; Riegel, R.; Hartmann, W.; Steiner, S.; Wardelmann, E.; Merkelbach-Bruse, S.; Tanaka, S.; Sonobe, H.; Schüle, R.; Buettner, R.; et al. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum. Pathol. 2011, 42, 1667–1675. [Google Scholar] [CrossRef]
- Reed, D.R.; Chawla, S.P.; Setty, B.; Mascarenhas, L.; Meyers, P.A.; Metts, J.; Harrison, D.J.; Lessnick, S.L.; Crompton, B.D.; Loeb, D.; et al. Phase 1 expansion trial of the LSD1 inhibitor seclidemstat (SP-2577) with and without topotecan and cyclophosphamide (TC) in patients (pts) with relapsed or refractory Ewing sarcoma (ES) and select sarcomas. J. Clin. Oncol. 2021, 39, TPS11577. [Google Scholar] [CrossRef]
- Parrish, J.K.; McCann, T.S.; Sechler, M.; Sobral, L.M.; Ren, W.H.; Jones, K.L.; Tan, A.C.; Jedlicka, P. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget 2018, 9, 33110–33123. [Google Scholar] [CrossRef] [PubMed]
- Karolak, M.; Tracy, I.; Shipley, J.; Walters, Z.S. Targeting EZH2 for the treatment of soft tissue sarcomas. J. Cancer Metastasis Treat. 2021, 7, 15. [Google Scholar] [CrossRef]
- Je, E.M.; An, C.H.; Yoo, N.J.; Lee, S.H. Mutational analysis of PIK3CA, JAK2, BRAF, FOXL2, IDH1, AKT1 and EZH2 oncogenes in sarcomas. Apmis 2012, 120, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.-W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Chi, S.N.; Yi, J.S.; Williams, P.M.; Roy-Chowdhuri, S.; Patton, D.R.; Coffey, B.; Reid, J.M.; Piao, J.; Saguilig, L.; Alonzo, T.A.; et al. Tazemetostat in patients with tumors with alterations in EZH2 or the SWI/SNF complex: Results from NCI-COG Pediatric MATCH trial Arm C (APEC1621C). J. Clin. Oncol. 2022, 40, 10009. [Google Scholar] [CrossRef]
- Jaboin, J.; Wild, J.; Hamidi, H.; Khanna, C.; Kim, C.J.; Robey, R.; Bates, S.E.; Thiele, C.J. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002, 62, 6108–6115. [Google Scholar]
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schäfer, B.W.; Burdach, S.; Richter, G.H. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2015, 7, 1451–1463. [Google Scholar] [CrossRef]
- Osgood, C.L.; Maloney, N.; Kidd, C.G.; Kitchen-Goosen, S.; Segars, L.; Gebregiorgis, M.; Woldemichael, G.M.; He, M.; Sankar, S.; Lessnick, S.L.; et al. Identification of Mithramycin Analogues with Improved Targeting of the EWS-FLI1 Transcription Factor. Clin. Cancer Res. 2016, 22, 4105–4118. [Google Scholar] [CrossRef]
- Caropreso, V.; Darvishi, E.; Turbyville, T.J.; Ratnayake, R.; Grohar, P.J.; McMahon, J.B.; Woldemichael, G.M. Englerin A Inhibits EWS-FLI1 DNA Binding in Ewing Sarcoma Cells. J. Biol. Chem. 2016, 291, 10058–10066. [Google Scholar] [CrossRef]
- Paronetto, M.P. Ewing Sarcoma Protein: A Key Player in Human Cancer. Int. J. Cell Biol. 2013, 2013, 642853. [Google Scholar] [CrossRef] [PubMed]
- Embree, L.J.; Azuma, M.; Hickstein, D.D. Ewing Sarcoma Fusion Protein EWSR1/FLI1 Interacts with EWSR1 Leading to Mitotic Defects in Zebrafish Embryos and Human Cell Lines. Cancer Res. 2009, 69, 4363–4371. [Google Scholar] [CrossRef] [PubMed]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS–FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.; Soldatenkov, V.; Dritschilo, A.; Notario, V. Poly(ADP-ribose) polymerase turnover alterations do not contribute to PARP overexpression in Ewing’s sarcoma cells. Oncol. Rep. 2002, 9, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 Inhibition as a Targeted Strategy to Treat Ewing’s Sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef]
- Chugh, R.; Ballman, K.V.; Helman, L.J.; Patel, S.; Whelan, J.S.; Widemann, B.; Lu, Y.; Hawkins, D.S.; Mascarenhas, L.; Glod, J.W.; et al. SARC025 arms 1 and 2: A phase 1 study of the poly(ADP-ribose) polymerase inhibitor niraparib with temozolomide or irinotecan in patients with advanced Ewing sarcoma. Cancer 2020, 127, 1301–1310. [Google Scholar] [CrossRef]
- Schafer, E.S.; Rau, R.; Berg, S.L.; Liu, X.; Minard, C.G.; Bishop, A.J.; Romero, J.C.; Hicks, M.J.; Nelson, M.D.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr. Blood Cancer 2019, 67, e28073. [Google Scholar] [CrossRef]
- Tanaka, K.; Suzuki, K.; Miyashita, K.; Wakasa, K.; Kawano, M.; Nakatsu, Y.; Tsumura, H.; Yoshida, M.A.; Oda, S. Activation of recombinational repair in Ewing sarcoma cells carrying EWS-FLI1 fusion gene by chromosome translocation. Sci. Rep. 2022, 12, 14764. [Google Scholar] [CrossRef]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Soler, M.; Morgado-Palacin, I.; Lafarga, V.; Lecona, E.; Murga, M.; Callen, E.; Azorin, D.; Alonso, J.; Lopez-Contreras, A.J.; Nussenzweig, A.; et al. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget 2016, 7, 58759–58767. [Google Scholar] [CrossRef]
- Koppenhafer, S.L.; Goss, K.L.; Terry, W.W.; Gordon, D.J. Inhibition of the ATR–CHK1 Pathway in Ewing Sarcoma Cells Causes DNA Damage and Apoptosis via the CDK2-Mediated Degradation of RRM2. Mol. Cancer Res. 2020, 18, 91–104. [Google Scholar] [CrossRef]
- Humme, D.; Haider, A.; Möbs, M.; Mitsui, H.; Suárez-Fariñas, M.; Ohmatsu, H.; Geilen, C.I.; Eberle, J.; Krueger, J.G.; Beyer, M.; et al. Aurora Kinase A Is Upregulated in Cutaneous T-Cell Lymphoma and Represents a Potential Therapeutic Target. J. Investig. Dermatol. 2015, 135, 2292–2300. [Google Scholar] [CrossRef]
- Winter, G.E.; Rix, U.; Lissat, A.; Stukalov, A.; Müllner, M.K.; Bennett, K.L.; Colinge, J.; Nijman, S.M.; Kubicek, S.; Kovar, H.; et al. An Integrated Chemical Biology Approach Identifies Specific Vulnerability of Ewing’s Sarcoma to Combined Inhibition of Aurora Kinases A and B. Mol. Cancer Ther. 2011, 10, 1846–1856. [Google Scholar] [CrossRef]
- Sourisseau, T.; Maniotis, D.; McCarthy, A.; Tang, C.; Lord, C.J.; Ashworth, A.; Linardopoulos, S. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol. Med. 2010, 2, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wei, Q.; Tan, L.; Li, Y.; Li, J.; Li, L.; Jiang, T.; Zhang, S.; Jin, H. Inhibition of AURKB, regulated by pseudogene MTND4P12, confers synthetic lethality to PARP inhibition in skin cutaneous melanoma. Am. J. Cancer Res. 2020, 10, 3458–3474. [Google Scholar] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef]
- Tirado, O.M.; Mateo-Lozano, S.; Villar, J.; Dettin, L.E.; Llort, A.; Gallego, S.; Ban, J.; Kovar, H.; Notario, V. Caveolin-1 (CAV1) Is a Target of EWS/FLI-1 and a Key Determinant of the Oncogenic Phenotype and Tumorigenicity of Ewing’s Sarcoma Cells. Cancer Res. 2006, 66, 9937–9947. [Google Scholar] [CrossRef]
- Niedan, S.; Kauer, M.; Aryee, D.N.T.; Kofler, R.; Schwentner, R.; Meier, A.; Pötschger, U.; Kontny, U.; Kovar, H. Suppression of FOXO1 is responsible for a growth regulatory repressive transcriptional sub-signature of EWS-FLI1 in Ewing sarcoma. Oncogene 2013, 33, 3927–3938. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Altaba, A.R.I. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Guo, W.; Tang, X.-D.; Tang, S.; Yang, Y. Preliminary report of combination chemotherapy including Arsenic trioxide for stage III osteosarcoma and Ewing sarcoma. Zhonghua Wai Ke Za Zhi (China J. Surg.) 2006, 44, 805–808. [Google Scholar] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Üren, A. GLI1 Is a Direct Transcriptional Target of EWS-FLI1 Oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Sherman, A.K.; Pawel, B.R. Expression of therapeutic targets in Ewing sarcoma family tumors. Hum. Pathol. 2012, 43, 1077–1083. [Google Scholar] [CrossRef]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2006, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Gierisch, M.E.; Pfistner, F.; Lopez-Garcia, L.A.; Harder, L.; Schäfer, B.W.; Niggli, F.K. Proteasomal Degradation of the EWS-FLI1 Fusion Protein Is Regulated by a Single Lysine Residue. J. Biol. Chem. 2016, 291, 26922–26933. [Google Scholar] [CrossRef]
- Ambati, S.R.; Lopes, E.C.; Kosugi, K.; Mony, U.; Zehir, A.; Shah, S.K.; Taldone, T.; Moreira, A.L.; Meyers, P.A.; Chiosis, G.; et al. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 2013, 8, 323–336. [Google Scholar] [CrossRef]
- Ma, Y.; Baltezor, M.; Rajewski, L.; Crow, J.; Samuel, G.; Staggs, V.S.; Chastain, K.M.; Toretsky, J.A.; Weir, S.J.; Godwin, A.K. Targeted inhibition of histone deacetylase leads to suppression of Ewing sarcoma tumor growth through an unappreciated EWS-FLI1/HDAC3/HSP90 signaling axis. J. Mol. Med. 2019, 97, 957–972. [Google Scholar] [CrossRef]
- Paiva, S.-L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef]
- Huang, S.; Xing, Y.; Liu, Y. Emerging roles for the ER stress sensor IRE1α in metabolic regulation and disease. J. Biol. Chem. 2019, 294, 18726–18741. [Google Scholar] [CrossRef] [PubMed]
- Wiese, W.; Siwecka, N.; Wawrzynkiewicz, A.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. IRE1α Inhibitors as a Promising Therapeutic Strategy in Blood Malignancies. Cancers 2022, 14, 2526. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Suehara, Y.; Kohsaka, S.; Hayashi, T.; Akaike, K.; Mukaihara, K.; Kurihara, T.; Kim, Y.; Okubo, T.; Ishii, M.; et al. IRE1α-XBP1 inhibitors exerted anti-tumor activities in Ewing’s sarcoma. Oncotarget 2018, 9, 14428–14443. [Google Scholar] [CrossRef]
- Li, S.; Yang, Q.; Wang, H.; Wang, Z.; Zuo, D.; Cai, Z.; Hua, Y. Prognostic significance of serum lactate dehydrogenase levels in Ewing’s sarcoma: A meta-analysis. Mol. Clin. Oncol. 2016, 5, 832–838. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, D.; De Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; Van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.-L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef]
- Altvater, B.; Kailayangiri, S.; Theimann, N.; Ahlmann, M.; Farwick, N.; Chen, C.; Pscherer, S.; Neumann, I.; Mrachatz, G.; Hansmeier, A.; et al. Common Ewing sarcoma-associated antigens fail to induce natural T cell responses in both patients and healthy individuals. Cancer Immunol. Immunother. 2014, 63, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.M.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2006, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.-Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Borowski, A.; van Valen, F.; Ulbrecht, M.; Weiss, E.H.; Blasczyk, R.; Jorgens, H.; Gobel, U.; Schneider, E.M. Monomorphic HLA class I-(non-A, non-B) expression on Ewing’s tumor cell lines, modulation by TNF-α and IFN-γ. Immunobiology 1999, 200, 1–20. [Google Scholar] [CrossRef]
- Thiel, U.; Schober, S.J.; Einspieler, I.; Kirschner, A.; Thiede, M.; Schirmer, D.; Gall, K.; Blaeschke, F.; Schmidt, O.; Jabar, S.; et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology 2017, 6, e1312239. [Google Scholar] [CrossRef]
- Blaeschke, F.; Thiel, U.; Kirschner, A.; Thiede, M.; Rubio, R.A.; Schirmer, D.; Kirchner, T.; Richter, G.H.; Mall, S.; Klar, R.; et al. Human HLA-A*02:01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T Cells specifically inhibit Ewing sarcoma growth in vitro and in vivo. Oncotarget 2016, 7, 43267–43280. [Google Scholar] [CrossRef]
- Schirmer, D.; Grünewald, T.G.P.; Klar, R.; Schmidt, O.; Wohlleber, D.; Rubio, R.A.; Uckert, W.; Thiel, U.; Bohne, F.; Busch, D.H.; et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. Oncoimmunology 2016, 5, e1175795. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2016, 6, e1250050. [Google Scholar] [CrossRef]
- Hsu, K.; Middlemiss, S.; Saletta, F.; Gottschalk, S.; McCowage, G.B.; Kramer, B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2020, 28, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wu, Z.; Luo, W. A Novel Treatment for Ewing’s Sarcoma: Chimeric Antigen Receptor-T Cell Therapy. Front. Immunol. 2021, 12, 707211. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Long, L.M.; Read, E.J.; Leitman, S.F.; Carter, C.S.; Tsokos, M.; Goletz, T.J.; Avila, N.; Berzofsky, J.A.; Helman, L.J.; et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med. Pediatr. Oncol. 2002, 38, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.D.; Hutchinson, R.J.; Hohenkirk, L.F.; McKenna, E.A.; Yanik, G.A.; Levine, J.; E Chang, A.; Braun, T.M.; Mulé, J.J. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001, 61, 8513–8519. [Google Scholar]
- Ghisoli, M.; Barve, M.; Schneider, R.; Mennel, R.; Lenarsky, C.; Wallraven, G.; Pappen, B.O.; LaNoue, J.; Kumar, P.; Nemunaitis, D.; et al. Pilot Trial of FANG Immunotherapy in Ewing’s Sarcoma. Mol. Ther. 2015, 23, 1103–1109. [Google Scholar] [CrossRef]
- Ghisoli, M.; Barve, M.; Mennel, R.; Lenarsky, C.; Horvath, S.; Wallraven, G.; Pappen, B.O.; Whiting, S.; Rao, D.; Senzer, N.; et al. Three-year Follow up of GMCSF/bi-shRNA furin DNA-transfected Autologous Tumor Immunotherapy (Vigil) in Metastatic Advanced Ewing’s Sarcoma. Mol. Ther. 2016, 24, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Caltavituro, A.; Buonaiuto, B.; Salomone, F.; Morra, R.; Pietroluongo, P.; De Placido, P.; Tortora, M.; Peddio, A.; Picozzi , F.; Ottaviano, M.; et al. Extraskeletal Ewing’s sarcoma of the mediastinum: Case report. Front. Oncol. 2023, 13, 1074378. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT | Target | Function in ES | Therapy | Status |
|---|---|---|---|---|
| NCT05275426 | Chk1 | DNA repair | Chk1 inhibitor (LY2880070) plus gemcitabine | ongoing |
| NCT03416517 | multi TK (VEGFR, PDGFR, C-KIT, FGFR) | Angiogenesis | Anlotinib | unknown |
| NCT04183062 | MARCKS (myristoylated alanine-rich C-kinase substrate) | Differentiation and tumor promoting function of cancer associated fibroblast (CAFs) | BIO-11006 | active, not recruiting |
| NCT05440786 | CDK4/6 | Cell proliferation | Abemaciclib | recruiting |
| NCT02243605 | multi TK (c-Met, VEGFR2, AXL e RET) | Proliferation and cell survival | Cabozantinib | active, non recruiting |
| NCT04901702 | PARP | DNA repair (after onivyde damage) | Onivyde + Talazoparib | recruiting |
| NCT05395741 | VEGFRs | Angiogenesis | Regorafenib | recruiting |
| NCT04890093 | HSP90 | Misfolding protein response | PEN-866 | not yet recruiting |
| (contains SN-38) | ||||
| NCT05093322 | VEGFR1, VEGFR2, VEGFR3, FGFR1, and CSF-1R | Cell survival and proliferation | Surufatinib | recruiting |
| NCT03373097 | GD-2 | Cell survival and proliferation | Anti-GD2 CAR T Cells | recruiting |
| NCT03715933 | DR5 | Cell death | INBRX-109 (DR5 agonist) | recruiting |
| NCT05159518 | CDK9 | Epigenetic and transcriptional reprogramming | PRT2527 | recruiting |
| (CDK9i) | ||||
| NCT04308330 | HDAC | Epigenetic and transcriptional reprogramming | Vorinostat | recruiting |
| NCT05071209 | ATR | DNA repair | Elimusertib | recruiting |
| NCT04195555 | IDH1/2 | Cell metabolism | Ivosidenib | recruiting |
| NCT04320888 | RET | Cell proliferation and survival | Selpercatinib | recruiting |
| NCT03698994 | ERK1/2 | Cell proliferation and survival | Ulixertinib | active not recruiting |
| NCT04284774 | farnesyltransferase inhibitor | Cell proliferation | Tipifarnib | recruiting |
| NCT04851119 | TBL1 (Transducin Beta Like Protein 1) | Cell proliferation and metastasization | Tegavivint | recruiting |
| NCT03213665 | EZH2 or the SWI/SNF complex | Epigenetic regulation | Tazemetostat | active not recruting |
| NCT03213652 | ALK and ROS1 | Proliferation and cell survival | Ensartinib | recruiting |
| NCT03213704 | NTRK | Proliferation and cell survival | Larotrectinib/Entrectinib | recruiting |
| NCT04897321 | B7H3 | Immune response | B7H3 CAR T Cell | recruiting |
| NCT03220035 | BRAFV600E | Proliferation and cell survival | Vemurafenib | active not recruiting |
| NCT03618381 | EGFR | Proliferation and cell survival | EGFR806 CAR T | recruiting |
| NCT03213678 | TSC or PI3K/mTOR | Proliferation and cell survival | Samotolisib | recruiting |
| NCT03425279 | AXL (RTK) | Proliferation and cell survival | BA3011 (CAB-AXL-ADC) | recruiting |
| NCT05182164 | TK and immune environment | Proliferation and cell survival and immunotherapy | Pembrolizumab + cabozantinib | recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caltavituro, A.; Buonaiuto, R.; Pietroluongo, E.; Morra, R.; Salomone, F.; De Placido, P.; Pagliuca, M.; Vaia, A.; Ottaviano, M.; Tortora, M.; et al. Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype. Biomedicines 2023, 11, 874. https://doi.org/10.3390/biomedicines11030874
Caltavituro A, Buonaiuto R, Pietroluongo E, Morra R, Salomone F, De Placido P, Pagliuca M, Vaia A, Ottaviano M, Tortora M, et al. Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype. Biomedicines. 2023; 11(3):874. https://doi.org/10.3390/biomedicines11030874
Chicago/Turabian StyleCaltavituro, Aldo, Roberto Buonaiuto, Erica Pietroluongo, Rocco Morra, Fabio Salomone, Pietro De Placido, Martina Pagliuca, Angelo Vaia, Margaret Ottaviano, Marianna Tortora, and et al. 2023. "Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype" Biomedicines 11, no. 3: 874. https://doi.org/10.3390/biomedicines11030874
APA StyleCaltavituro, A., Buonaiuto, R., Pietroluongo, E., Morra, R., Salomone, F., De Placido, P., Pagliuca, M., Vaia, A., Ottaviano, M., Tortora, M., De Placido, S., Palmieri, G., & Giuliano, M. (2023). Shifting from a Biological-Agnostic Approach to a Molecular-Driven Strategy in Rare Cancers: Ewing Sarcoma Archetype. Biomedicines, 11(3), 874. https://doi.org/10.3390/biomedicines11030874