

The Role of ERα and ERβ in Castration-Resistant Prostate Cancer and Current Therapeutic Approaches

 ,
, 


Abstract
1. Introduction
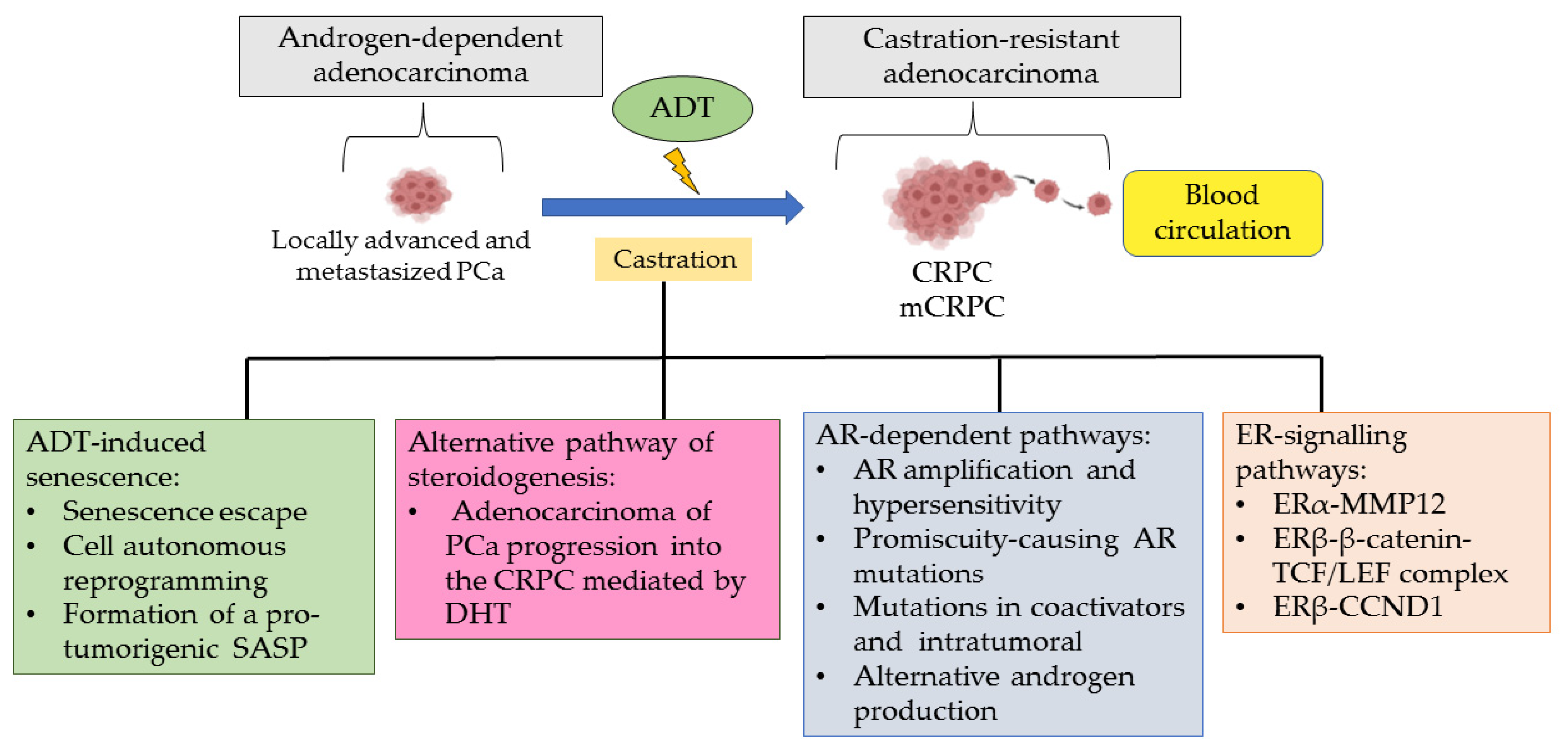
2. Role of ADT in Developing CRPC
3. Roles of ERα and ERβ in Castration-Resistant Prostate Cancer
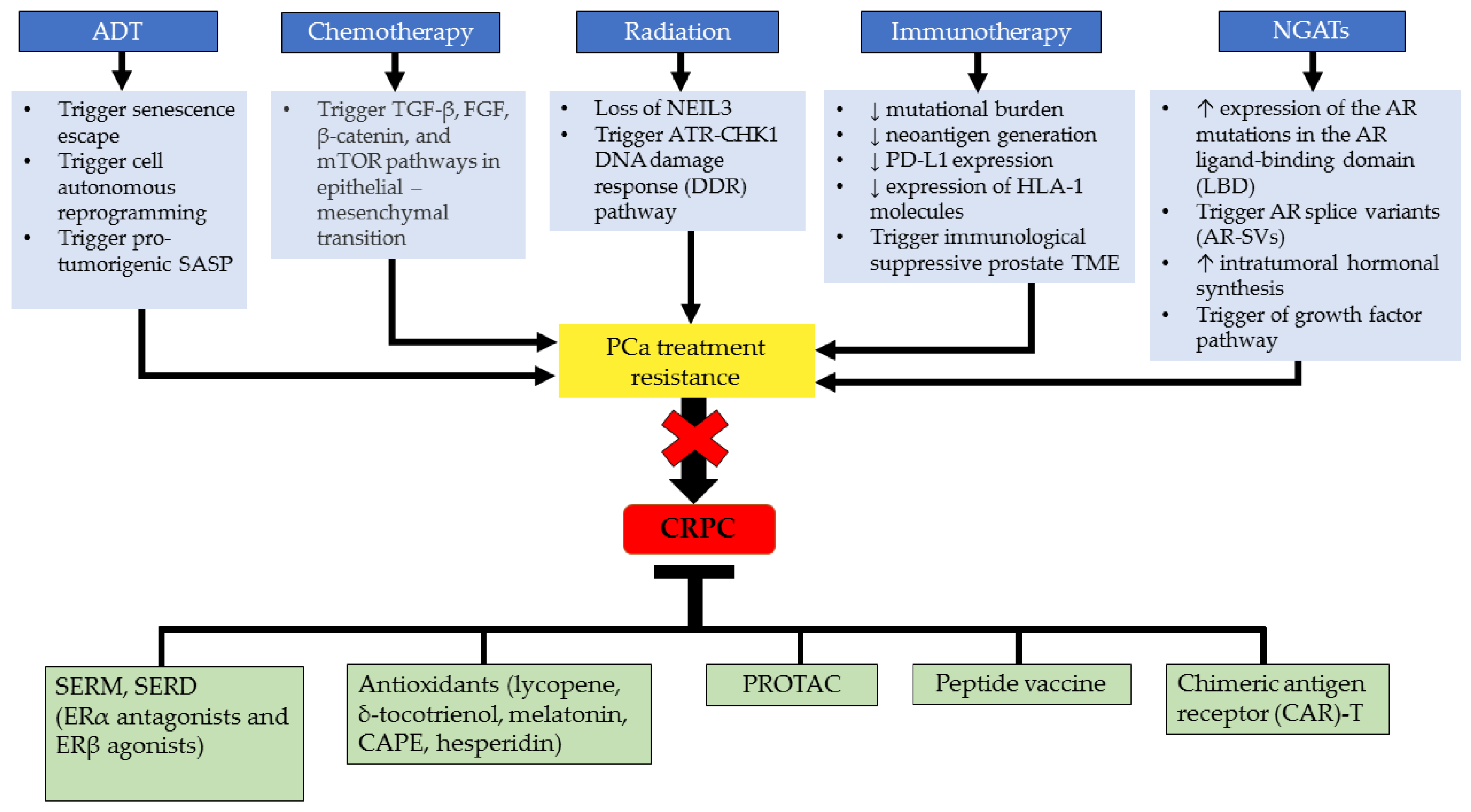
4. Current CRPC Treatment and Challenges
5. Molecular Targeted Therapeutic Approaches in Mitigating CRPC
5.1. Drugs Targeting Estrogen Related to CRPC Treatment
5.2. Anti-Estrogen Therapies
5.3. Selective Inhibitors
5.4. Antioxidants
5.5. Advanced Immunotherapy Approach
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| 3β-Adiol | 5α-Androstane-3β,17β-Diol |
| AA | Abiraterone acetate |
| ADT | Androgen deprivation therapy |
| AE | Adverse events |
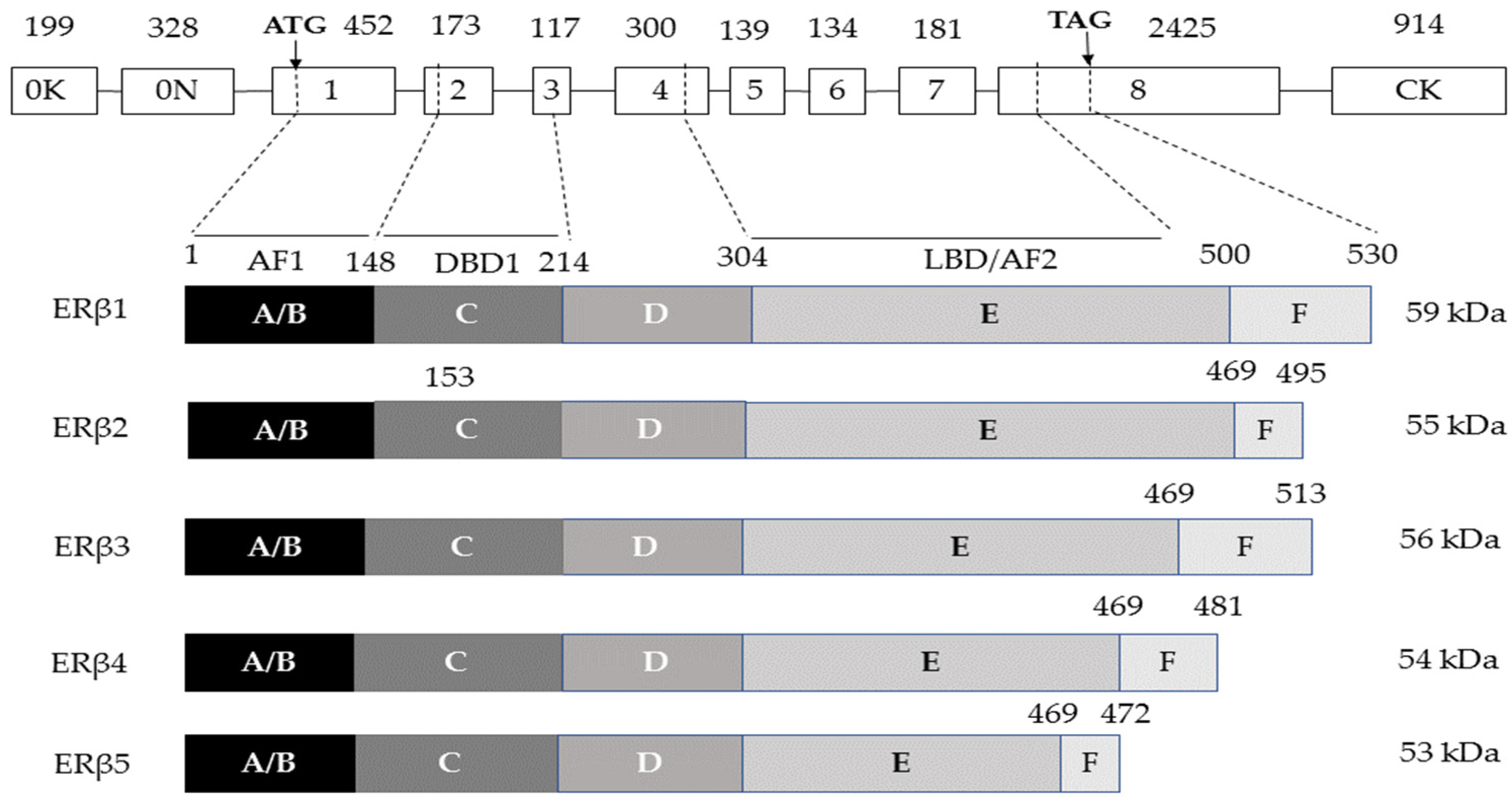
| AF1 | Activation function 1 |
| AF2 | Activation function 2 |
| AR | Androgen receptor |
| AR-SV | Androgen receptor-splice variant |
| ATP | Adenosine triphosphate |
| BAD | Bcl-2-associated death |
| BF | Biochemical failure |
| BMS | Bone marrow stromal |
| CAPE | Caffeic acid phenethyl ester |
| CAR-T cell | Chimeric antigen receptor- t cell |
| CCND1 | Cyclin D1 |
| CES1 | Carboxylesterase 1 |
| CHD1 | Chromodomain-helicase DNA-binding protein |
| CRPC | Castration-resistant prostate cancer |
| CYP17A1 | Cytochrome P450 17A1 |
| CYP191A1 | Cytochrome P450 191A1 |
| DBD | DNA-binding domain |
| DDR | DNA damage response |
| DHEA | Dehydroepiandrosterone |
| DHEA-S | Dehydroepiandrosterone sulfate |
| DHT | Dihydrotestosterone |
| DNA | Deoxyribonucleic acid |
| DPN | Diarylprepionitrile |
| DSBs | Double-strand breaks |
| E2 | Estradiol |
| EMT | Epithelial-mesenchymal transition |
| ENZ | Enzalutamide |
| ER | Estrogen receptor |
| EREs | Estrogen response elements |
| ERR | Estrogen-related receptor |
| ERα | Estrogen receptor alpha |
| ERβ | Estrogen receptor beta |
| ESR1 | Estrogen receptor 1 |
| ESR2 | Estrogen receptor 2 |
| FGF | Fibroblast growth factor |
| FOX | Forkhead box protein |
| FOXO | Forehead box O |
| GEM | Genetically engineered mouse |
| GnRH | Gonadotropin-releasing hormone |
| HIF-α | Hypoxia-inducible factor 1-alpha |
| HLA | Human leukocyte antigen |
| ICIs | Immune checkpoint inhibitors |
| KO | Knocked-out |
| LBD | Ligand-binding domain |
| LD | Lipid droplet |
| LH | Luteinizing hormone |
| LHRH | Luteinizing hormone-releasing hormone |
| mCRPC | Metastasis castration-resistant prostate cancer |
| MDSC | Myeloid-derived suppressor cells |
| mHSPC | Metastatic hormone-sensitive prostate cancer |
| MMP12 | Matrix metalloproteinase 12 |
| mRNA | Messenger ribonucleic acid |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor-kappa beta |
| NGATs | Next-generation anti-androgen therapies |
| OS | Overall survival |
| PAP | Prostatic acid phosphatase |
| PCa | Prostate cancer |
| PCSLC | Prostate cancer stem-like cell |
| PDC | Poorly differentiated carcinoma |
| PDE | Patient-derived explant |
| PDKI | Phosphorylation of phosphoinositide-dependent kinase |
| PDO | Patient-derived organoids |
| PDX | Patient-derived xenografts |
| PDX | Patient-derived xenograft |
| PDXOs | Patient-derived xenograft-derived organoids |
| PFS | Progression-free survival |
| p-mTOR | Phosphorylated mTOR |
| PPT | Propylpyrazoletriol |
| PPV | Personalized peptide vaccination |
| PROTAC | Proteolysis targeting chimeras |
| PSA | Prostate-specific antigen |
| PSCA | Prostate stem cell antigen |
| PSMA | Prostate-specific membrane antigen |
| PTEN | Phosphatase and tensin homolog |
| RECIST | Response evaluation criteria in solid tumors |
| RNA | Ribonucleic acid |
| RP | Radical prostatectomy |
| SASP | Senescence-associated secretory phenotype |
| SERDs | Selective estrogen receptor down regulators |
| SERMs | Selective estrogen receptor modulators |
| SPOP | Speckle-type poz protein |
| SRC | Steroid receptor coactivator |
| SRC-PI3K-AKT-mTOR | Steroid receptor coactivator/phosphoinositide 3-kinase/akt/mammalian target of rapamycin |
| SSBs | Single-strand breaks |
| STEAP | Six-transmembrane prostate epithelial antigen |
| TCF/LEF | T cell factor/lymphoid enhancer factor transcription factors |
| TGF-β | Transforming growth factor-beta |
| TME | The tumour microenvironment |
| TRAMP | Transgenic adenocarcinoma of the mouse prostate |
| VCaP | Vertebral cancer of the human prostate |
| VEGF-A | Vascular endothelial growth factor A |
| WLS | Wntless |
| δ-TT | Delta-tocotrienol |
References
- Ferro, M.; de Cobelli, O.; Musi, G.; del Giudice, F.; Carrieri, G.; Busetto, G.M.; Falagario, U.G.; Sciarra, A.; Maggi, M.; Crocetto, F.; et al. Radiomics in Prostate Cancer: An up-to-Date Review. Ther. Adv. Urol. 2022, 14, 17562872221109020. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.M.; Gellen, L.P.A.; Leal, D.F.D.V.B.; Pastana, L.F.; Vinagre, L.W.M.S.; Aquino, V.T.; Fernandes, M.R.; de Assumpção, P.P.; Burbano, R.M.R.; dos Santos, S.E.B.; et al. Correlation between Genomic Variants and Worldwide Epidemiology of Prostate Cancer. Genes 2022, 13, 1039. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Prim. 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of Resistance in Castration-Resistant Prostate Cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365. [Google Scholar] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.R.; Chen, Y.A.; Kao, W.H.; Lai, C.H.; Lin, H.; Hsieh, J.T. Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease. Biomedicines 2022, 10, 1872. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers 2019, 11, 1418. [Google Scholar] [CrossRef]
- Fujimura, T.; Takayama, K.; Takahashi, S.; Inoue, S. Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine. Road Nanomed. Precis. Med. 2020, 901–929. [Google Scholar]
- Nelles, J.L.; Hu, W.Y.; Prins, G.S. Estrogen Action and Prostate Cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 437–451. [Google Scholar] [CrossRef]
- Tong, D. Selective Estrogen Receptor Modulators Contribute to Prostate Cancer Treatment by Regulating the Tumor Immune Microenvironment. J. Immunother. Cancer 2022, 10, 2944. [Google Scholar] [CrossRef]
- Cipolletti, M.; Fernandez, V.S.; Montalesi, E.; Marino, M.; Fiocchetti, M. Beyond the Antioxidant Activity of Dietary Polyphenols in Cancer: The Modulation of Estrogen Receptors (ERs) Signaling. Int. J. Mol. Sci. 2018, 19, 2624. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen Deprivation Therapy: Progress in Understanding Mechanisms of Resistance and Optimizing Androgen Depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate Cancer Progression after Androgen Deprivation Therapy: Mechanisms of Castrate Resistance and Novel Therapeutic Approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The Androgen Receptor Fuels Prostate Cancer by Regulating Central Metabolism and Biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Rove, K.O.; Crawford, E.D. Androgen Annihilation as a New Therapeutic Paradigm in Advanced Prostate Cancer. Curr. Opin. Urol. 2013, 23, 208–213. [Google Scholar] [CrossRef]
- Nguyen, C.; Lairson, D.R.; Swartz, M.D.; Du, X.L. Risks of Major Long-Term Side Effects Associated with Androgen-Deprivation Therapy in Men with Prostate Cancer. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 999–1009. [Google Scholar] [CrossRef]
- Desai, K.; McManus, J.M.; Sharifi, N. Hormonal Therapy for Prostate Cancer. Endocr. Rev. 2021, 42, 354–373. [Google Scholar] [CrossRef]
- Choi, J.B.; Koh, J.S. LHRH Agonist and Antagonist for Prostate Cancer. Manag. Adv. Prostate Cancer 2018, 127–132. [Google Scholar]
- Boland, J.; Choi, W.; Lee, M.; Lin, J. Cardiovascular Toxicity of Androgen Deprivation Therapy. Curr. Cardiol. Rep. 2021, 23, 109. [Google Scholar] [CrossRef]
- Gamat, M.; McNeel, D.G. Androgen Deprivation and Immunotherapy for the Treatment of Prostate Cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Morris, M.J.; Rumble, R.B.; Milowsky, M.I. Optimizing Anticancer Therapy in Metastatic Non-Castrate Prostate Cancer: ASCO Clinical Practice Guideline Summary. J. Oncol. Pract. 2018, 14, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.P.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone Acetate plus Prednisone in Patients with Newly Diagnosed High-Risk Metastatic Castration-Sensitive Prostate Cancer (LATITUDE): Final Overall Survival Analysis of a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juárez, S.; Merseburger, A.S.; Özguroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 661. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Lin, P.; Tombal, B.; Saad, F.; Higano, C.S.; Joshua, A.M.; Parli, T.; Rosbrook, B.; van Os, S.; Beer, T.M. Five-Year Survival Prediction and Safety Outcomes with Enzalutamide in Men with Chemotherapy-Naïve Metastatic Castration-Resistant Prostate Cancer from the PREVAIL Trial. Eur. Urol. 2020, 78, 347–357. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Saylor, P.J.; Smith, M.R. Treatment and Prevention of Bone Complications from Prostate Cancer. Bone 2011, 48, 88–95. [Google Scholar] [CrossRef]
- Mohamad, N.V.; Ima-Nirwana, S.; Chin, K.Y. The Effects of Annatto Tocotrienol on Body Composition and Serum Adiponectin, Leptin and Glucose Level in a Rat Model of Androgen Deficiency Induced by Buserelin. Med. Health 2019, 14, 168–179. [Google Scholar]
- Yusoff, N.A.; Taib, I.S.; Budin, S.B.; Mohamed, M. Paternal Fenitrothion Exposures in Rats Causes Sperm Dna Fragmentation in F0 and Histomorphometric Changes in Selected Organs of F1 Generation. Toxics 2021, 9, 159. [Google Scholar] [CrossRef]
- Othman, H.; Yamin, A.H.A.; Isa, N.M.; Bahadzor, B.; Zakaria, S.Z.S. Diagnostic performance of prostate health index (PHI) in predicting prostate cancer on prostate biopsy. Malays. J. Pathol. 2020, 42, 209–214. [Google Scholar]
- Ko, C.C.; Yeh, L.R.; Kuo, Y.T.; Chen, J.H. Imaging Biomarkers for Evaluating Tumor Response: RECIST and Beyond. Biomark. Res. 2021, 9, 52. [Google Scholar] [CrossRef]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular Determinants of Resistance to Antiandrogen Therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Patel, B.B.; Autorino, R.; Smith, S.C.; Gewirtz, D.A.; Saleh, T. Senescence and Castration Resistance in Prostate Cancer: A Review of Experimental Evidence and Clinical Implications. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188424. [Google Scholar] [CrossRef]
- Araujo, J.C.; Mathew, P.; Armstrong, A.J.; Braud, E.L.; Posadas, E.; Lonberg, M.; Gallick, G.E.; Trudel, G.C.; Paliwal, P.; Agrawal, S.; et al. Dasatinib Combined with Docetaxel for Castration-Resistant Prostate Cancer: Results from a Phase 1-2 Study. Cancer 2012, 118, 63–71. [Google Scholar] [CrossRef]
- Shamhari, A.; Abd Hamid, Z.; Budin, S.B.; Shamsudin, N.J.; Taib, I.S. Bisphenol A and Its Analogues Deteriorate the Hormones Physiological Function of the Male Reproductive System: A Mini-Review. Biomedicines 2021, 9, 1744. [Google Scholar] [CrossRef]
- Lombardi, A.P.G.; Vicente, C.M.; Porto, C.S. Estrogen Receptors Promote Migration, Invasion and Colony Formation of the Androgen-Independent Prostate Cancer Cells PC-3 through β-Catenin Pathway. Front. Endocrinol. 2020, 11, 184. [Google Scholar] [CrossRef]
- Jefferi, N.E.S.E.S.; Shamhari, A.; Hamid, Z.A.A.; Budin, S.B.B.; Zulkifly, A.M.Z.M.Z.; Roslan, F.N.N.; Taib, I.S.S. Knowledge Gap in Understanding the Steroidogenic Acute Regulatory Protein Regulation in Steroidogenesis Following Exposure to Bisphenol A and Its Analogues. Biomedicines 2022, 10, 1281. [Google Scholar] [CrossRef] [PubMed]
- Lafront, C.; Germain, L.; Weidmann, C.; Audet-Walsh, É. A Systematic Study of the Impact of Estrogens and Selective Estrogen Receptor Modulators on Prostate Cancer Cell Proliferation. Sci. Rep. 2020, 10, 4024. [Google Scholar] [CrossRef] [PubMed]
- Ellem, S.J.; Schmitt, J.F.; Pedersen, J.S.; Frydenberg, M.; Risbridger, G.P. Local Aromatase Expression in Human Prostate Is Altered in Malignancy. J. Clin. Endocrinol. Metab. 2004, 89, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.J.; Ellem, S.J.; Patchev, V.; Fritzemeier, K.-H.; Risbridger, G.P. The Role of ERα and ERβ in TheProstate: Insights from Genetic Models and Isoform-Selective Ligands. Tissue-Specific Estrogen Action Nov. Mech. Nov. Ligands Nov. Ther. 2007, 2006, 131–148. [Google Scholar]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Cooke, P.S.; Mesa, A.M.; Sirohi, V.K.; Levin, E.R. Role of Nuclear and Membrane Estrogen Signaling Pathways in the Male and Female Reproductive Tract. Differentiation 2021, 118, 24–33. [Google Scholar] [CrossRef]
- Gustafsson, J.A.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. Mol. Biol. 2019, 191, 105312. [Google Scholar] [CrossRef] [PubMed]
- Nabha, S.M.; Burck dos Santos, E.; Yamamoto, H.A.; Belizi, A.; Dong, Z.; Meng, H.; Saliganan, A.; Sabbota, A.; Daniel Bonfil, R.; Cher, M.L. Bone Marrow Stromal Cells Enhance Prostate Cancer Cell Invasion through Type I Collagen in an MMP-12 Dependent Manner. Wiley Online Libr. 2008, 122, 11. [Google Scholar] [CrossRef] [PubMed]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar] [PubMed]
- Qu, L.G.; Wardan, H.; Davis, I.D.; Iddawela, M.; Sluka, P.; Pezaro, C.J. Circulating Oestrogen Receptor Mutations and Splice Variants in Advanced Prostate Cancer. Wiley Online Libr. 2019, 124, 50–56. [Google Scholar] [CrossRef]
- Grindstad, T.; Skjefstad, K.; Andersen, S.; Ness, N.; Nordby, Y.; Al-Saad, S.; Fismen, S.; Donnem, T.; Khanehkenari, M.R.; Busund, L.T.; et al. Estrogen Receptors α and β and Aromatase as Independent Predictors for Prostate Cancer Outcome. Sci. Rep. 2016, 6, 33114. [Google Scholar] [CrossRef]
- Nelson, A.W.; Tilley, W.D.; Neal, D.E.; Carroll, J.S. Estrogen Receptor Beta in Prostate Cancer: Friend or Foe? Endocr. Relat. Cancer 2014, 21, T219–T234. [Google Scholar] [CrossRef]
- Alam, S.; Zunic, A.; Venkat, S.; Feigin, M.E.; Atanassov, B.S. Regulation of Cyclin D1 Degradation by Ubiquitin-Specific Protease 27X Is Critical for Cancer Cell Proliferation and Tumor Growth. Mol. Cancer Res. 2022, 20, OF1–OF12. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Yang, C.J.; Lin, C.Y.; Lee, Y.S.; Wu, J.M. Control of Stability of Cyclin D1 by Quinone Reductase 2 in CWR22Rv1 Prostate Cancer Cells. Carcinogenesis 2012, 33, 670–677. [Google Scholar] [CrossRef]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.Å. Mechanisms of Estrogen Action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen Receptor Mutations in Human Disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef]
- Poola, I.; Abraham, J.; Baldwin, K.; Saunders, A.; Bhatnagar, R. Estrogen Receptors Beta4 and Beta5 Are Full Length Functionally Distinct ERβ Isoforms: Cloning from Human Ovary and Functional Characterization. Endocrine 2005, 27, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Lam, H.M.; Wu, S.; Song, D.; Levin, L.; Cheng, L.; Wu, C.L.; Ho, S.M. Estrogen Receptor Β2 and Β5 Are Associated with Poor Prognosis in Prostate Cancer, and Promote Cancer Cell Migration and Invasion. Endocr. Relat. Cancer 2010, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Božovićbožović, A.; Mandušić, V.M.; Todorović, L.T.; Krajnović, M.K. Estrogen Receptor Beta: The Promising Biomarker and Potential Target in Metastases. Int. J. Mol. Sci. 2021, 22, 1656. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta Impedes Prostate Cancer EMT by Destabilizing HIF-1alpha and Inhibiting VEGF-Mediated Snail Nuclear Localization: Implications for Gleason Grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc Promotes Therapeutic Resistance Development of Neuroendocrine Prostate Cancer by Differentially Regulating MiR-421/ATM Pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef]
- Zellweger, T.; Stürm, S.; Rey, S.; Zlobec, I.; Gsponer, J.R.; Rentsch, C.A.; Terracciano, L.M.; Bachmann, A.; Bubendorf, L.; Ruiz, C. Estrogen Receptor β Expression and Androgen Receptor Phosphorylation Correlate with a Poor Clinical Outcome in Hormone-Naïve Prostate Cancer and Are Elevated in Castration-Resistant Disease. Endocr. Relat. Cancer 2013, 20, 403–413. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; D’Abronzo, L.S.; Lou, W.; Evans, C.P.; Gao, A.C. Wntless Promotes Cellular Viability and Resistance to Enzalutamide in Castration-Resistant Prostate Cancer Cells. Am. J. Clin. Exp. Urol. 2019, 7, 203. [Google Scholar]
- Shapiro, D.; Tareen, B. Current and Emerging Treatments in the Management of Castration-Resistant Prostate Cancer. Expert Rev. Anticancer Ther. 2012, 12, 951–964. [Google Scholar] [CrossRef]
- Chen, S.; Li, W.; Guo, A. LOXL1-AS1 Predicts Poor Prognosis and Promotes Cell Proliferation, Migration, and Invasion in Osteosarcoma. Biosci. Rep. 2019, 39, BSR20190447. [Google Scholar] [CrossRef] [PubMed]
- Van Dodewaard-De Jong, J.M.; Verheul, H.M.W.; Bloemendal, H.J.; De Klerk, J.M.H.; Carducci, M.A.; Van Den Eertwegh, A.J.M. New Treatment Options for Patients With Metastatic Prostate Cancer: What Is The Optimal Sequence? Clin. Genitourin. Cancer 2015, 13, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Felizola, S.J.A.; Kurotaki, Y.; Fujishima, F.; McNamara, K.M.; Suzuki, T.; Arai, Y.; Sasano, H. Cyclin D1 (CCND1) Expression Is Involved in Estrogen Receptor Beta (ERβ) in Human Prostate Cancer. Prostate 2012, 73, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Alfano, A.; Xu, J.; Yang, X.; Deshmukh, D.; Qiu, Y. SRC Kinase-Mediated Tyrosine Phosphorylation of TUBB3 Regulates Its Stability and Mitotic Spindle Dynamics in Prostate Cancer Cells. Pharmaceutics 2022, 14, 932. [Google Scholar] [CrossRef]
- Liang, Z.; Cao, J.; Tian, L.; Shen, Y.; Yang, X.; Lin, Q.; Zhang, R.; Liu, H.; Du, X.; Shi, J.; et al. Aromatase-Induced Endogenous Estrogen Promotes Tumour Metastasis through Estrogen Receptor-α/Matrix Metalloproteinase 12 Axis Activation in Castration-Resistant Prostate Cancer. Cancer Lett. 2019, 467, 72–84. [Google Scholar] [CrossRef]
- Lacroix, M.; Leclercq, G. Relevance of Breast Cancer Cell Lines as Models for Breast Tumours: An Update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef]
- Ghallab, A. In Vitro Test Systems and Their Limitations. EXCLI J. 2013, 12, 1024. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Adamiecki, R.; Hryniewicz-Jankowska, A.; Ortiz, M.A.; Li, X.; Porter-Hansen, B.A.; Nsouli, I.; Bratslavsky, G.; Kotula, L. In Vivo Models for Prostate Cancer Research. Cancers 2022, 14, 5321. [Google Scholar] [CrossRef]
- Mai, C.-W.; Chin, K.-Y.; Foong, L.-C.; Pang, K.-L.; Yu, B.; Shu, Y.; Chen, S.; Cheong, S.-K.; Chua, C.W. Modeling Prostate Cancer: What Does It Take to Build an Ideal Tumor Model? Cancer Lett. 2022, 543, 215794. [Google Scholar] [CrossRef] [PubMed]
- Ślusarz, A.; Jackson, G.A.; Day, J.K.; Shenouda, N.S.; Bogener, J.L.; Browning, J.D.; Fritsche, K.L.; MacDonald, R.S.; Besch-Williford, C.L.; Lubahn, D.B. Aggressive Prostate Cancer Is Prevented in ERαKO Mice and Stimulated in ERβKO TRAMP Mice. Endocrinology 2012, 153, 4160–4170. [Google Scholar] [CrossRef]
- Risbridger, G.; Wang, H.; Young, P.; Kurita, T.; Wong, Y.Z.; Lubahn, D.; Gustafsson, J.-A.; Cunha, G. Evidence That Epithelial and Mesenchymal Estrogen Receptor-α Mediates Effects of Estrogen on Prostatic Epithelium. Dev. Biol. 2001, 229, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Abugomaa, A.; Elbadawy, M.; Yamawaki, H.; Usui, T.; Sasaki, K. Emerging Roles of Cancer Stem Cells in Bladder Cancer Progression, Tumorigenesis, and Resistance to Chemotherapy: A Potential Therapeutic Target for Bladder Cancer. Cells 2020, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, M.; Abugomaa, A.; Yamawaki, H.; Usui, T.; Sasaki, K. Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research. Cancers 2020, 12, 777. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, A.; Lafront, C.; Peillex, C.; Pelletier, M.; Audet-Walsh, É. Impacts of Endocrine-Disrupting Chemicals on Prostate Function and Cancer. Environ. Res. 2022, 204, 112085. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical Models of Prostate Cancer: Patient-Derived Xenografts, Organoids, and Other Explant Models. Cold Spring Harb. Perspect. Med. 2018, 8, a030536. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Clark, A.K.; Porter, L.H.; Toivanen, R.; Bakshi, A.; Lister, N.L.; Pook, D.; Pezaro, C.J.; Sandhu, S.; Keerthikumar, S.; et al. The MURAL Collection of Prostate Cancer Patient-Derived Xenografts Enables Discovery Through Preclinical Models of Uro-Oncology. Nat. Commun. 2021, 12, 5049. [Google Scholar] [CrossRef]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of Androgen Receptor Splice Variant-7 (AR-V7) in Prostate Cancer Resistance to 2nd-Generation Androgen Receptor Signaling Inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic Characterization of a Unique Neuroendocrine Transdifferentiation Prostate Circulating Tumor Cell-Derived EXplant Model. Nat. Commun. 2020, 11, 1884. [Google Scholar] [CrossRef]
- Van Hemelryk, A.; Tomljanovic, I.; Stuurman, D.; de Ridder, C.M.A.; Teubel, W.J.; Erkens-Schulze, S.; van de Werken, H.J.G.; van Royen, M.; Grudniewska, M.; Jenster, G.W.; et al. Patient-Derived Xenografts and Organoids Recapitulate Castration-Resistant Prostate Cancer with Sustained Androgen Receptor Signaling. Eur. J. Cancer 2022, 174, S43. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Sinnwell, J.P.; Thompson, K.J.; Copland, J.A.; Marlow, L.A.; Miller, J.L.; Yin, P.; Gao, B. Establishing and Characterizing Patient-Derived Xenografts Using Pre-Chemotherapy Percutaneous Biopsy and Post-Chemotherapy Surgical Samples from a Prospective Neoadjuvant Breast Cancer Study. Breast Cancer Res. 2017, 19, 130. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Asemota, S.; Schwartzberg, L.S.; Guestini, F.; McNamara, K.M.; Pierobon, M.; Font-Tello, A.; Qiu, X.; Xie, Y.; Rao, P.K.; et al. Androgen Receptor Is a Non-Canonical Inhibitor of Wild-Type and Mutant Estrogen Receptors in Hormone Receptor-Positive Breast Cancers. iScience 2019, 21, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Coussy, F.; Lavigne, M.; de Koning, L.; El Botty, R.; Nemati, F.; Naguez, A.; Bataillon, G.; Ouine, B.; Dahmani, A.; Montaudon, E. Response to MTOR and PI3K Inhibitors in Enzalutamide-Resistant Luminal Androgen Receptor Triple-Negative Breast Cancer Patient-Derived Xenografts. Theranostics 2020, 10, 1531–1543. [Google Scholar] [CrossRef]
- Gomez, L.; Kovac, J.R.; Lamb, D.J. CYP17A1 Inhibitors in Castration-Resistant Prostate Cancer. Steroids 2015, 95, 80–87. [Google Scholar] [CrossRef]
- Lohiya, V.; Aragon-Ching, J.B.; Sonpavde, G. Role of Chemotherapy and Mechanisms of Resistance to Chemotherapy in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10, CMO-S34535. [Google Scholar] [CrossRef]
- Feng, J.; Byrne, N.M.; Al Jamal, W.; Coulter, J.A. Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development. Cancers 2019, 11, 1989. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Yang, J.; Peng, S.; Zhou, Q.; Yao, K.; Cai, W.; Xie, Z.; Qin, F.; Li, H. Loss of NEIL3 Activates Radiotherapy Resistance in the Progression of Prostate Cancer. Cancer Biol. Med. 2022, 19, 1193–1210. [Google Scholar] [CrossRef]
- Mohler, M.L.; Sikdar, A.; Ponnusamy, S.; Hwang, D.-J.; He, Y.; Miller, D.D.; Narayanan, R. An Overview of Next-Generation Androgen Receptor-Targeted Therapeutics in Development for the Treatment of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 2124. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K. The Evolving Landscape of Immunotherapy in Advanced Prostate Cancer. Immunotherapy 2019, 11, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Fortis, S.P.; Perez, S.A. Prostate Cancer: Any Room Left for Immunotherapies? Immunotherapy 2019, 11, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bujanda, Z.; Drake, C.G. Myeloid-derived Cells in Prostate Cancer Progression: Phenotype and Prospective Therapies. J. Leukoc. Biol. 2017, 102, 393–406. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E. IL-23 Secreted by Myeloid Cells Drives Castration-Resistant Prostate Cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A Novel Second-Line Treatment for Metastatic Castration-Resistant Prostate Cancer. Drug Des. Devel. Ther. 2011, 5, 117–124. [Google Scholar] [PubMed]
- Xu, P.; Wasielewski, L.J.; Yang, J.C.; Cai, D.; Evans, C.P.; Murphy, W.J.; Liu, C. The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines 2022, 10, 1778. [Google Scholar] [CrossRef]
- Formenti, A.M.; Dalla Volta, A.; di Filippo, L.; Berruti, A.; Giustina, A. Effects of Medical Treatment of Prostate Cancer on Bone Health. Trends Endocrinol. Metab. 2021, 32, 135–158. [Google Scholar] [CrossRef]
- Jiménez, N.; Reig, Ò.; Marín-Aguilera, M.; Aversa, C.; Ferrer-Mileo, L.; Font, A.; Rodriguez-Vida, A.; Climent, M.Á.; Cros, S.; Chirivella, I.; et al. Transcriptional Profile Associated with Clinical Outcomes in Metastatic Hormone-Sensitive Prostate Cancer Treated with Androgen Deprivation and Docetaxel. Cancers 2022, 14, 4757. [Google Scholar] [CrossRef]
- Damiano, J.S.; Wasserman, E. Molecular Pathways: Blockade of the PRLR Signaling Pathway as a Novel Antihormonal Approach for the Treatment of Breast and Prostate Cancer. Clin. Cancer Res. 2013, 19, 1644–1650. [Google Scholar] [CrossRef]
- Smith, K.; Galazi, M.; Openshaw, M.R.; Wilson, P.; Sarker, S.J.; O’Brien, N.; Alifrangis, C.; Stebbing, J.; Shamash, J. The Use of Transdermal Estrogen in Castrate-Resistant, Steroid-Refractory Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, e217–e223. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H. Selective Estrogen Receptor Modulators (SERMS): Keys to Understanding Their Function. Menopause 2020, 27, 1171–1176. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Nuti, R. Selective Estrogen Receptor Modulator (SERM) for the Treatment of Osteoporosis in Postmenopausal Women: Focus on Lasofoxifene. Clin. Interv. Aging 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; Condorelli, R.A.; Barbagallo, F.; La Vignera, S.; Calogero, A.E. Endocrinology of the Aging Prostate: Current Concepts. Front. Endocrinol. 2021, 12, 554078. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I.; Mathew, S.; Rahman, S. Recent Progress in Selective Estrogen Receptor Downregulators (SERDs) for the Treatment of Breast Cancer. RSC Med. Chem. 2020, 11, 438–454. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Feng, F.Y.; Wang, Y.; Cao, X.; Han, S.; Wilder-Romans, K.; Navone, N.M.; Logothetis, C.; Taichman, R.S.; Keller, E.T.; et al. Mechanistic Support for Combined MET and AR Blockade in Castration-Resistant Prostate Cancer. Neoplasia 2016, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.H.; Nunez-Nateras, R.; Hou, Y.X.; Bryce, A.H.; Northfelt, D.W.; Dueck, A.C.; Wong, B.; Stanton, M.L.; Joseph, R.W.; Castle, E.P. A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, 196–202.e1. [Google Scholar] [CrossRef]
- Shazer, R.L.; Jain, A.; Galkin, A.V.; Cinman, N.; Nguyen, K.N.; Natale, R.B.; Gross, M.; Green, L.; Bender, L.I.; Holden, S.; et al. Raloxifene, an Oestrogen-Receptor-β-Targeted Therapy, Inhibits Androgen-Independent Prostate Cancer Growth: Results from Preclinical Studies and a Pilot Phase II Clinical Trial. BJU Int. 2006, 97, 691–697. [Google Scholar] [CrossRef]
- Stein, S.; Zoltick, B.; Peacock, T.; Holroyde, C.; Haller, D.; Armstead, B.; Malkowicz, S.B.; Vaughn, D.J. Phase II Trial of Toremifene in Androgen-Independent Prostate Cancer: A Penn Cancer Clinical Trials Group Trial. Am. J. Clin. Oncol. 2001, 24, 283–285. [Google Scholar] [CrossRef]
- Hariri, W.; Sudha, T.; Bharali, D.J.; Cui, H.; Mousa, S.A. Nano-Targeted Delivery of Toremifene, an Estrogen Receptor-α Blocker in Prostate Cancer. Pharm. Res. 2015, 32, 2764–2774. [Google Scholar] [CrossRef]
- Chadha, M.K.; Ashraf, U.; Lawrence, D.; Tian, L.; Levine, E.; Silliman, C.; Escott, P.; Payne, V.; Trump, D.L. Phase II Study of Fulvestrant (Faslodex) in Castration Resistant Prostate Cancer. Prostate 2008, 68, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, J.; Kaulfub, S.; Jarry, H.; Bremmer, F.; Stettner, M.; Burfeind, P.; Thelen, P. Prospects of Estrogen Receptor β Activation in the Treatment of Castration-Resistant Prostate Cancer. Oncotarget 2017, 8, 34971. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Tran, J.D.; Muldong, M.T.; Nollet, E.A.; Schulz, V.V.; Jensen, C.C.; Hazlehurst, L.A.; Corey, E.; Durden, D.; Jamieson, C.; et al. Hypoxia-Induced PIM Kinase and Laminin-Activated Integrin A6 Mediate Resistance to PI3K Inhibitors in Bone-Metastatic CRPC. Am. J. Clin. Exp. Urol. 2019, 7, 297. [Google Scholar] [PubMed]
- Cham, J.; Venkateswaran, A.R.; Bhangoo, M. Targeting the PI3K-AKT-MTOR Pathway in Castration Resistant Prostate Cancer: A Review Article. Clin. Genitourin. Cancer 2021, 19, 563-e1. [Google Scholar] [CrossRef]
- Lilis, I.; Giopanou, I.; Papadaki, H.; Gyftopoulos, K. The Expression of P-MTOR and COUP-TFII Correlates with Increased Lymphangiogenesis and Lymph Node Metastasis in Prostate Adenocarcinoma. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2018; Volume 36, pp. 311.e27–311.e35. [Google Scholar]
- Barata, P.C.; Magi-Galluzzi, C.; Gupta, R.; Dreicer, R.; Klein, E.A.; Garcia, J.A. Association of MTOR Pathway Markers and Clinical Outcomes in Patients with Intermediate-/High-Risk Prostate Cancer: Long-Term Analysis. Clin. Genitourin. Cancer 2019, 17, 366–372. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M. Phase II Trial of the PI3 Kinase Inhibitor Buparlisib (BKM-120) with or without Enzalutamide in Men with Metastatic Castration Resistant Prostate Cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.-W.; Swales, K.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef]
- Semenas, J.; Wang, T.; Sajid Syed Khaja, A.; Firoj Mahmud, A.K.M.; Simoulis, A.; Grundström, T.; Fällman, M.; Persson, J.L. Targeted Inhibition of ERα Signaling and PIP5K1α/Akt Pathways in Castration-resistant Prostate Cancer. Mol. Oncol. 2021, 15, 968. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN LossIpatasertib in Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I. A Phase I Dose-Escalation Study of Enzalutamide in Combination with the AKT Inhibitor AZD5363 (Capivasertib) in Patients with Metastatic Castration-Resistant Prostate Cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L. A Phase II Study of the Dual MTOR Inhibitor MLN0128 in Patients with Metastatic Castration Resistant Prostate Cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Percent, I.J.; Babu, S.; Cultrera, J.; Mehlhaff, B.A.; Goodman, O.B.; Morris, D.; Schnadig, I.D.; Albany, C.; Shore, N.D. Phase 1b/2 Study of Enzalutamide (ENZ) with LY3023414 (LY) or Placebo (PL) in Patients (Pts) with Metastatic Castration-Resistant Prostate Cancer (MCRPC) after Progression on Abiraterone. J. Clin. Oncol. 2019, 37, 5009. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.P.G.; Cavalheiro, R.P.; Porto, C.S.; Vicente, C.M. Estrogen Receptor Signaling Pathways Involved in Invasion and Colony Formation of Androgen-Independent Prostate Cancer Cells PC-3. Int. J. Mol. Sci. 2021, 22, 1153. [Google Scholar] [CrossRef]
- Singla, R.K.; Sai, C.S.; Chopra, H.; Behzad, S.; Bansal, H.; Goyal, R.; Gautam, R.K.; Tsagkaris, C.; Joon, S.; Singla, S.; et al. Natural Products for the Management of Castration-Resistant Prostate Cancer: Special Focus on Nanoparticles Based Studies. Front. Cell Dev. Biol. 2021, 9, 745177. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Singla, R.K.; Capasso, A. Multifaceted Effects of Lycopene: A Boulevard to the Multitarget-Based Treatment for Cancer. Molecules 2021, 26, 5333. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Fang, W.K.; Yang, S.M.; Wu, M.C.; Chen, T.J.; Chen, C.M.; Lin, T.Y.; Liu, K.L.; Wu, C.M.; Chen, Y.C.; et al. Natural Product Myricetin Is a Pan-KDM4 Inhibitor Which with Poly Lactic-Co-Glycolic Acid Formulation Effectively Targets Castration-Resistant Prostate Cancer. J. Biomed. Sci. 2022, 29, 29. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Parmakhtiar, B.; Simoneau, A.R.; Xie, J.; Fruehauf, J.; Lilly, M.; Zi, X. Lycopene Enhances Docetaxel’s Effect in Castration-Resistant Prostate Cancer Associated with Insulin-like Growth Factor I Receptor Levels. Neoplasia 2011, 13, 108. [Google Scholar] [CrossRef]
- Fontana, F.; Moretti, R.M.; Raimondi, M.; Marzagalli, M.; Beretta, G.; Procacci, P.; Sartori, P.; Montagnani Marelli, M.; Limonta, P. δ-Tocotrienol Induces Apoptosis, Involving Endoplasmic Reticulum Stress and Autophagy, and Paraptosis in Prostate Cancer Cells. Cell Prolif. 2019, 52, e12576. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, C.; Yang, X.; Liu, L.; Hu, J.; Hou, Y.; Tao, H.; Sugimura, H.; Chen, Z.; Wang, L.; et al. Melatonin Inhibits Lipid Accumulation to Repress Prostate Cancer Progression by Mediating the Epigenetic Modification of CES1. Clin. Transl. Med. 2021, 11, e449. [Google Scholar] [CrossRef]
- Chuu, C.P.; Lin, H.P.; Ciaccio, M.F.; Kokontis, J.M.; Hause, R.J.; Hiipakka, R.A.; Liao, S.; Jones, R.B. Caffeic Acid Phenethyl Ester Suppresses the Proliferation of Human Prostate Cancer Cells through Inhibition of P70S6K and Akt Signaling Networks. Cancer Prev. Res. 2012, 5, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.P.; Jiang, S.S.; Chuu, C.P. Caffeic Acid Phenethyl Ester Causes P21Cip1 Induction, Akt Signaling Reduction, and Growth Inhibition in PC-3 Human Prostate Cancer Cells. PLoS ONE 2012, 7, e31286. [Google Scholar] [CrossRef] [PubMed]
- McEleny, K.; Coffey, R.; Morrissey, C.; Fitzpatrick, J.M.; Watson, R.W.G. Caffeic Acid Phenethyl Ester-Induced PC-3 Cell Apoptosis Is Caspase-Dependent and Mediated through the Loss of Inhibitors of Apoptosis Proteins. BJU Int. 2004, 94, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Dallner, G.; Bentinger, M.; Hussain, S.; Sinha, I.; Yang, J.; Schwank-Xu, C.; Zheng, X.; Swiezewska, E.; Brismar, K.; Valladolid-Acebes, I. Dehydro-Tocotrienol-β Counteracts Oxidative-Stress-Induced Diabetes Complications in Db/Db Mice. Antioxidants 2021, 10, 1070. [Google Scholar] [CrossRef]
- Jang, W.Y.; Kim, M.-Y.; Cho, J.Y. Antioxidant, Anti-Inflammatory, Anti-Menopausal, and Anti-Cancer Effects of Lignans and Their Metabolites. Int. J. Mol. Sci. 2022, 23, 15482. [Google Scholar] [CrossRef] [PubMed]
- AV, S.; AH, H. Phytoestrogens and Their Effects. Eur. J. Pharmacol. 2014, 741, 230–236. [Google Scholar]
- Jeong, S.A.; Yang, C.; Song, J.; Song, G.; Jeong, W.; Lim, W. Hesperidin Suppresses the Proliferation of Prostate Cancer Cells by Inducing Oxidative Stress and Disrupting Ca2+ Homeostasis. Antioxidants 2022, 11, 1633. [Google Scholar] [CrossRef]
- Patra, P.; Bhattacharya, M.; Sharma, A.R.; Ghosh, P.; Sharma, G.; Patra, B.C.; Mallick, B.; Lee, S.S.; Chakraborty, C. Identification and Design of a Next-Generation Multi Epitopes Bases Peptide Vaccine Candidate Against Prostate Cancer: An In Silico Approach. Cell Biochem. Biophys. 2020, 78, 495–509. [Google Scholar] [CrossRef]
- Sun, B.L. Immunotherapy in Treatment of Metastatic Prostate Cancer: An Approach to Circumvent Immunosuppressive Tumor Microenvironment. Prostate 2021, 81, 1125–1134. [Google Scholar] [CrossRef]
- Park, Y.H.; Jung, A.R.; Kim, G.E.; Kim, M.Y.; Sung, J.W.; Shin, D.; Cho, H.J.; Ha, U.S.; Hong, S.H.; Kim, S.W.; et al. GV1001 Inhibits Cell Viability and Induces Apoptosis in Castration-Resistant Prostate Cancer Cells through the AKT/NF-Κb/VEGF Pathway. J. Cancer 2019, 10, 6269–6277. [Google Scholar] [CrossRef]
- Noguchi, M.; Fujimoto, K.; Arai, G.; Uemura, H.; Matsumoto, H.; Kohjimoto, Y.; Nakatsu, H.; General, A.; Takenaka, H.A.; Naito, S.; et al. Personalized Peptide Vaccination for Castration-Resistant Prostate Cancer Progressing after Docetaxel: A Randomized, Double-Blind, Placebo-Controlled, Phase III. arXiv 2020. [Google Scholar] [CrossRef]
- Lebraud, H.; Heightman, T.D. Protein Degradation: A Validated Therapeutic Strategy with Exciting Prospects. Essays Biochem. 2017, 61, 517–527. [Google Scholar] [PubMed]
- Liu, L.; Shi, L.; Wang, Z.; Zeng, J.; Wang, Y.; Xiao, H.; Zhu, Y. Targeting Oncoproteins for Degradation by Small Molecule-Based Proteolysis-Targeting Chimeras (PROTACs) in Sex Hormone-Dependent Cancers. Front. Endocrinol. 2022, 13, 839857. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An Oral Androgen Receptor PROTAC Degrader for Prostate Cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Snyder, L.B.; Neklesa, T.K.; Chen, X.; Dong, H.; Ferraro, C.; Gordon, D.A.; Macaluso, J.; Pizzano, J.; Wang, J.; Willard, R.R.; et al. Discovery of ARV-110, a First in Class Androgen Receptor Degrading PROTAC for The Treatment of Men with Metastatic Castration Resistant Prostate Cancer. Cancer Res. 2021, 81 (Suppl. 13), 43. [Google Scholar] [CrossRef]
- Ma, Q.; Gomes, E.M.; Lo, A.S.Y.; Junghans, R.P. Advanced Generation Anti-Prostate Specific Membrane Antigen Designer T Cells for Prostate Cancer Immunotherapy. Prostate 2014, 74, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef]
- Xiao, L.; Luo, Y.; Tai, R.; Zhang, N. Estrogen Receptor β Suppresses Inflammation and the Progression of Prostate Cancer. Mol. Med. Rep. 2019, 49, 3555–3563. [Google Scholar] [CrossRef]
- Schade, G.R.; Holt, S.K.; Zhang, X.; Song, D.; Wright, J.L.; Zhao, S.; Kolb, S.; Lam, H.-M.; Levin, L.; Leung, Y.-K.; et al. Prostate cancer expression profiles of cytoplasmic ERβ1 and nuclear ERβ2 are associated with poor outcomes following radical prostatectomy. J. Urol. 2016, 195, 1760–1766. [Google Scholar] [CrossRef]
- Christoforou, P.; Christopoulos, P.F.; Koutsilieris, M. The Role of Estrogen Receptor β in Prostate Cancer. Mol. Med. 2014, 20, 427. [Google Scholar] [CrossRef]
- Ramírez-de-Arellano, A.; Pereira-Suárez, A.L.; Rico-Fuentes, C.; López-Pulido, E.I.; Villegas-Pineda, J.C.; Sierra-Diaz, E. Distribution and Effects of Estrogen Receptors in Prostate Cancer: Associated Molecular Mechanisms. Front. Endocrinol. 2021, 12, 811578. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef] [PubMed]
- Misawa, A.; Inoue, S. Estrogen-Related Receptors in Breast Cancer and Prostate Cancer. Front. Endocrinol. 2015, 6, 83. [Google Scholar] [CrossRef]
- Leach, D.A.; Powell, S.M.; Bevan, C.L. WOMEN IN CANCER THEMATIC REVIEW: New Roles for Nuclear Receptors in Prostate Cancer. Endocr. Relat. Cancer 2016, 23, T85–T108. [Google Scholar] [CrossRef]
- Nelson, A.W.; Groen, A.J.; Miller, J.L.; Warren, A.Y.; Holmes, K.A.; Tarulli, G.A.; Tilley, W.D.; Katzenellenbogen, B.S.; Hawse, J.R.; Gnanapragasam, V.J.; et al. Comprehensive Assessment of Estrogen Receptor Beta Antibodies in Cancer Cell Line Models and Tissue Reveals Critical Limitations in Reagent Specificity. Mol. Cell. Endocrinol. 2017, 440, 138–150. [Google Scholar] [CrossRef]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramström, M.; Söderberg, O.; et al. Insufficient Antibody Validation Challenges Oestrogen Receptor Beta Research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.D.; Liu, X.; Zuo, F.; Eisenbraun, T.L.; Wiley, S.R.; Kraus, R.J.; Mertz, J.E. Estrogen-Related Receptor Alpha 1 Functionally Binds as a Monomer to Extended Half-Site Sequences Including Ones Contained within Estrogen-Response Elements. Mol. Endocrinol. 1997, 11, 342–352. [Google Scholar] [PubMed]
- Fradet, A.; Bouchet, M.; Delliaux, C.; Gervais, M.; Kan, C.; Benetollo, C.; Pantano, F.; Vargas, G.; Bouazza, L.; Croset, M.; et al. Estrogen Related Receptor Alpha in Castration-Resistant Prostate Cancer Cells Promotes Tumor Progression in Bone. Oncotarget 2016, 7, 77071. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Wu, D.; Wang, S.; Chen, Z.; Xiang, S.; Chan, F.L. Orphan Nuclear Receptors as Regulators of Intratumoral Androgen Biosynthesis in Castration-Resistant Prostate Cancer. Oncogene 2021, 40, 2625–2634. [Google Scholar] [CrossRef]
- Yu, S.; Wong, Y.C.; Wang, X.H.; Ling, M.T.; Ng, C.F.; Chen, S.; Chan, F.L. Orphan Nuclear Receptor Estrogen-Related Receptor-Beta Suppresses in Vitro and in Vivo Growth of Prostate Cancer Cells via P21(WAF1/CIP1) Induction and as a Potential Therapeutic Target in Prostate Cancer. Oncogene 2008, 27, 3313–3328. [Google Scholar] [CrossRef]
- Pugach, E.K.; Blenck, C.L.; Dragavon, J.M.; Langer, S.J.; Leinwand, L.A. Estrogen Receptor Profiling and Activity in Cardiac Myocytes. Mol. Cell. Endocrinol. 2016, 431, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.F.; Wang, L.; Spetsieris, N.; Boukovala, M.; Efstathiou, E.; Brössner, C.; Warner, M.; Gustafsson, J.A. Estrogen Receptor β and Treatment with a Phytoestrogen Are Associated with Inhibition of Nuclear Translocation of EGFR in the Prostate. Proc. Natl. Acad. Sci. USA 2021, 118, e2011269118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study Model | Estrogen Receptor | Findings | Author |
|---|---|---|---|
| Human PC-3 cell line | ERβ |
ERβ regulates the cell cycle of PCa by controlling the expression of CCND1 (p < 0.05) | [63] |
| Tissue microarray consisting of PCa samples from CRPC patients (n = 101) |
ERα ERβ |
ERα
↓ ERβ1 ↑ (p < 0.05) | [57] |
| Human PC-3 cell line | ERα | ERα ↑ | [37] |
| Human PC-3 and LNCaP cell lines | ERβ1 | ERβ1 and 3β-Adiol repress mesenchymal characteristics VEGF-A and EMT expression ↓ (p < 0.05) TGF-β and hypoxia reduced expression of ERβ1 | [55] |
| Blood-derived RNA samples from CRPC patients (n = 42) |
ERα ERβ |
Detection of four mutations of ERα (E380Q, L536Q, Y537S and D538G) ERβ splice variant concentrations ↓ | [45] |
| Public genomic datasets from patients with metastatic or advanced prostate cancer (n= 150) and patients with early prostate cancer (n = 492) |
ERα ERβ | The prevalence of ERα and ERβ mutations:
| [85] |
| Primary tumor tissue from radical prostatectomy (RP) patients (n = 535) | ERβ | ERβ was correlated with decreased time to biochemical failure (BF) (p = 0.002) | [46] |
| Human PC-3 (derived from bone metastasis) and DU-145 (derived from brain metastasis) cell lines |
ERα ERβ | ERα activation via PPT (ERα-selective agonist) ERβ activation via DPN (ERβ-selective agonist) Both ERα and ERβ increase the migration and invasion of PC-3 cell lines | [35] |
| Treatment | Number (n) | Findings | Author |
|---|---|---|---|
| ADT+ toremifene 300–640 mg/m2 | 15 | No cancer inhibitory effect | [109] |
| Raloxifene 60 mg | 13 | Partial effect (5 of 13 patients) | [108] |
| Fulvestrant 500 mg, 250 mg | 20 | No patients reduced PSA by >50% | [111] |
| Raloxifene (60 mg) + bicaltamide (50 mg) | 18 | Partial effect (4 of 18 patients) | [107] |
| Concentrations of 5 µM and 25 µM of ERβ-selective agonist, 8-VE2, and 8β-VE2 in VCaP cells | n/a | The expression of AR protein ↓ by 52% AR mRNA ↓ by 40% | [112] |
| Nano-targeted delivery of toremifene (1.0 to 100 μM) in PC3M cancer cell line | n/a | Prostate tumor growth ↓ by blocking ERα | [110] |
| Treatment | Number (n) | Findings | Author |
|---|---|---|---|
| Tamoxifen (20 mg/kg) + ISA-2011B (20 mg/kg) in PC-3 cells | 25 | ERα expression significantly ↓ (p < 0.001) | [124] |
| 1 M of PI3K specific inhibitors (Wortmannin), 200 nm of AKT inhibitor (MK2206) and 5 nM of SRC-family kinase inhibitor (PP2) | n/a | Size and number of colony of PC-3 cell line significantly ↓ (p < 0.05) | [125] |
| mTOR inhibitor (MLN0128) | 9 | 8 of 9 patients quit taking the medication before the endpoint and experienced Grade 3 adverse effects, such as mucositis, rash, pain, delirium, and dyspnea | [122] |
| Pan-PI3K inhibitor (BKM120) +/− enzalutamide | The median progression-free survival was 1.9 months (in combination with enzalutamide) as opposed to 3.5 months | [117] | |
| Akt inhibitor (AZD5363) + enzalutamide in mCRPC | 13 | 3 of 13 patients (23%) experienced a PSA decrease of more than 50% Patients experienced side effects: rash (20%) and hyperglycemia (26.7%) | [121] |
| Treatment | Findings | Author |
|---|---|---|
| Lycopene | Growth inhibition on DU145 cells ↑ Docetaxel + lycopene: tumor regression ↑, with a 38% increase in antitumor efficacy (p < 0.05) | [129] |
| δ-Tocotrienol | δ-TT exerts a cytotoxic/proapoptotic activity in CRPC cells | [130] |
| Melatonin | Inhibition of tumor growth CES1 expression ↑ | [131] |
| CAPE | Proliferation of DU-145 and PC-3 cells dose-dependently ↓ | [132] |
| CAPE | Proliferation of DU-145 and PC-3 cells dose-dependently ↓ | [133] |
| CAPE | Induced apoptosis in PC-3 | [134] |
| Hesperidin | Suppressed the proliferation of androgen-independent PC-3 and DU145 Improved the anticancer effects of the chemotherapy drug cisplatin in PC-3 and DU145 cells | [138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jefferi, N.E.S.; Shamhari, A.‘A.; Noor Azhar, N.K.Z.; Shin, J.G.Y.; Kharir, N.A.M.; Azhar, M.A.; Hamid, Z.A.; Budin, S.B.; Taib, I.S. The Role of ERα and ERβ in Castration-Resistant Prostate Cancer and Current Therapeutic Approaches. Biomedicines 2023, 11, 826. https://doi.org/10.3390/biomedicines11030826
Jefferi NES, Shamhari A‘A, Noor Azhar NKZ, Shin JGY, Kharir NAM, Azhar MA, Hamid ZA, Budin SB, Taib IS. The Role of ERα and ERβ in Castration-Resistant Prostate Cancer and Current Therapeutic Approaches. Biomedicines. 2023; 11(3):826. https://doi.org/10.3390/biomedicines11030826
Chicago/Turabian StyleJefferi, Nur Erysha Sabrina, Asma’ ‘Afifah Shamhari, Nur Khayrin Zulaikha Noor Azhar, Joyce Goh Yi Shin, Nur Annisa Mohd Kharir, Muhammad Afiq Azhar, Zariyantey Abd Hamid, Siti Balkis Budin, and Izatus Shima Taib. 2023. "The Role of ERα and ERβ in Castration-Resistant Prostate Cancer and Current Therapeutic Approaches" Biomedicines 11, no. 3: 826. https://doi.org/10.3390/biomedicines11030826
APA StyleJefferi, N. E. S., Shamhari, A. ‘A., Noor Azhar, N. K. Z., Shin, J. G. Y., Kharir, N. A. M., Azhar, M. A., Hamid, Z. A., Budin, S. B., & Taib, I. S. (2023). The Role of ERα and ERβ in Castration-Resistant Prostate Cancer and Current Therapeutic Approaches. Biomedicines, 11(3), 826. https://doi.org/10.3390/biomedicines11030826