Abstract
Background: Atrial fibrillation (AF) is promoted by various stimuli like angiotensin II, endothelin-1, epinephrine/norepinephrine, vagal activation, or mechanical stress, all of which activate receptors coupled to G-proteins of the Gαq/Gα11-family (Gq). Besides pro-fibrotic and pro-inflammatory effects, Gq-mediated signaling induces inositol trisphosphate receptor (IP3R)-mediated intracellular Ca2+ mobilization related to delayed after-depolarisations and AF. However, direct evidence of arrhythmogenic Gq-mediated signaling is absent. Methods and results: To define the role of Gq in AF, transgenic mice with tamoxifen-inducible, cardiomyocyte-specific Gαq/Gα11-deficiency (Gq-KO) were created and exposed to intracardiac electrophysiological studies. Baseline electrophysiological properties, including heart rate, sinus node recovery time, and atrial as well as AV nodal effective refractory periods, were comparable in Gq-KO and control mice. However, inducibility and mean duration of AF episodes were significantly reduced in Gq-KO mice—both before and after vagal stimulation. To explore underlying mechanisms, left atrial cardiomyocytes were isolated from Gq-KO and control mice and electrically stimulated to study Ca2+-mobilization during excitation–contraction coupling using confocal microscopy. Spontaneous arrhythmogenic Ca2+ waves and sarcoplasmic reticulum content-corrected Ca2+ sparks were less frequent in Gq-KO mice. Interestingly, nuclear but not cytosolic Ca2+ transient amplitudes were significantly decreased in Gq-KO mice. Conclusion: Gq-signaling promotes arrhythmogenic atrial Ca2+-release and AF in mice. Targeting this pathway, ideally using Gq-selective, biased receptor ligands, may be a promising approach for the treatment and prevention of AF. Importantly, the atrial-specific expression of the Gq-effector IP3R confers atrial selectivity mitigating the risk of life-threatening ventricular pro-arrhythmic effects.
1. Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia. As well as causing debilitating symptoms, AF is associated with considerable morbidity and increased mortality []. Current strategies to treat AF—both antiarrhythmic drugs as well as catheter ablation—are of moderate efficacy only. Moreover, while catheter ablation is associated with a certain procedural risk, antiarrhythmic drugs are generally not well tolerated and often have to be discontinued because of substantial side effects, including potentially life-threatening ventricular pro-arrhythmic effects [].
Numerous underlying pathological entities and conditions promote AF through neurohumoral triggers []. Many of these stimuli activate receptors coupled to G-proteins of the Gq-family (Gq) defined by the α-subunit isoforms Gαq and Gα11. In fact, pro-arrhythmic effects have been consistently demonstrated for the predominant cardiac Gq-coupled receptors, namely the angiotensin II receptor type 1 (AT1 receptor), the endothelin-1 receptor A (ETA receptor), the M3 muscarinic acetylcholine receptor (M3 receptor) and the alpha-1 adrenergic receptor []. In addition, immediate as well as chronic responses to mechanical forces can promote arrhythmia []. Interestingly, we could recently demonstrate that Gq can form a functional mechanosignaling complex with Piezo1 []. However, even though abundant evidence points to a central role of Gq-mediated signal transduction in AF, direct evidence of Gq-mediated arrhythmogenic effects and the putative mechanisms is absent [].
Ectopic activity, particularly within the pulmonary veins, can act as a trigger on a vulnerable atrial substrate and as a driver maintaining AF. Delayed after-depolarisations (DADs) constitute the most important mechanism of ectopic activity in AF. It has been shown that diastolic Ca2+ leak from the sarcoplasmic reticulum leads to an increased inward current via the Na+-Ca2+-exchanger is the underlying cause of DADs in patients with AF [,,]. Ca2+ release from the sarcoplasmic reticulum is regulated by ryanodine receptors (RyR2) as well as a second set of Ca2+ release channels, the inositol 1,4,5-trisphosphate (IP3) receptors. A large body of evidence demonstrates that type 2 IP3 receptors (IP3R) facilitate arrhythmogenic Ca2+ leak and AF-related ectopic activity []. IP3R is activated by IP3 in response to Gq-mediated signaling via phospholipase C. Of note, the expression and function of IP3R, but not of RyR2, are enhanced in AF []. From a pharmacological standpoint, it is most intriguing that IP3R expression in atrial myocytes is 6- to 10-fold higher than in ventricular myocytes and that IP3R-mediated electrophysiological effects on Ca2+ homeostasis are absent in ventricular myocytes [,]. As the use of all currently approved antiarrhythmic drugs is limited by potentially life-threatening ventricular pro-arrhythmic effects, this renders the Gq-IP3R-signaling pathway a promising target for the treatment of AF.
Here we investigate possible arrhythmogenic mechanisms and effects of Gq-mediated signaling in the context of AF, as well as its suitability as a pharmacological target.
2. Methods
2.1. Conditional Cardiomyocyte-Specific Gαq/Gα11-Deficient Mice
Mice with a tamoxifen-inducible, cardiomyocyte-specific Gαq/Gα11-deficiency (Gq-KO) were kindly provided by Prof. Nina Wettschureck, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany. Briefly, those Gq-KO mice harbor floxed Gnaq and Gna11−/− alleles as well as a tamoxifen-inducible Cre recombinase under the promoter of the mouse αMHC (MYH6) gene (MHCCreERT2) as previously reported [,]. Cre-mediated recombination of floxed alleles was induced by intraperitoneal injection of 1 mg tamoxifen dissolved in 50 μL miglyol oil on 5 consecutive days in 8-week-old Gq-KO mice. MHCCreERT2; GnaqWT/WT; Gna11+/+ mice served as control group and likewise underwent the tamoxifen-induction protocol. Experiments were performed 2 weeks after the end of induction.
All animal experiments were approved by the responsible federal authority (LAGeSo Berlin, approval TVA G0006/18) and performed conforming to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Reporting in the manuscript follows the recommendations in the ARRIVE guidelines.
2.2. Invasive Electrophysiological Studies in Mice
For electrophysiological studies, mice were anesthetized with isoflurane (1.6 vol.% isoflurane/air) and placed on a heated surgical pad to maintain a constant body temperature. Limb electrodes were inserted subcutaneously to record a 6-lead surface ECG. After hair removal, a midline cervical incision was made, and the right jugular vein exposed to introduce a 2-French Octapolar diagnostic catheter (CIBermouse cath; NuMed, Inc., Cross Roads, TX, USA) connected to a digital electrophysiology recording system (EP Tracer, CardioTek, Maastricht, The Netherlands). The distal tip of the catheter was positioned in the right ventricle in a way that enabled recording of ventricular electrograms with the distal electrodes and atrial electrograms with the proximal electrodes. Inducibility of AF was determined before and two minutes after intraperitoneal injection of 50 ng/g carbachol (Sigma-Aldrich) by programmed electrical stimulation according to a murine AF model previously described by Wakimoto et al. []. AF was defined as the occurrence of fragmented atrial electrograms with irregular cycle lengths below 25 ms and absolute ventricular arrhythmia for at least 1 s. Animals were subsequently euthanized with a lethal dose of isoflurane followed by cervical dislocation.
2.3. Animal In Vitro Experiments
All chemicals and reagents were obtained from Sigma-Aldrich (St Louis, MO, USA) unless noted otherwise. Tyrode solution contained (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 Dglucose, 10 Hepes; pH 7.4 with NaOH. Atrial cardiomyocytes were isolated from WT and Gq-KO mice (n = 5 animals / group) as previously described using enzymatic digestion []. Cells were subsequently loaded with Fluo-4-AM (Thermo Fisher, Waltham, MA, USA), and [Ca2+]-related fluorescence was measured using confocal line-scan imaging (Zeiss LSM 800, excitation at 488 nm, emission collected at > 515 nm) []. Experiments were performed at 35°C in Tyrode solution (3 mM [Ca2+]), and Ca2+ transients were elicited using electrical field stimulation (1 Hz). Longitudinal scan lines were chosen, and the cellular nucleus visually identified and included when feasible. Line scan images were used to derive cytosolic and nuclear Ca2+ transient release and removal characteristics (i.e., peak fluorescence: F/F0; TF50: time to 50% of maximal Ca2+ release; TAU: decay constant of Ca2+ transient) []. Changes in [Ca]i in intact paced myocytes are expressed as an F/F0 where F represents cellular Fluo-4 fluorescence and F0 is diastolic Fluo-4 fluorescence. Ca2+ waves and Ca2+ spark frequencies were measured during a resting period upon stop of electrical stimulation at steady-state (i.e., a minimum of two minutes electrical stimulation) and manually quantified in a blinded fashion. Ca2+ wave propagation velocity was measured as previously described []. Ca2+ spark parameters were analyzed using the automated ImageJ Plugin SparkMaster []. SR Ca2+ content was assessed in longitudinal line scans after Ca2+ spark/wave measurements using Caffeine (20 mM) evoked transients []. Analysis of Ca2+ transients was performed using ImageJ and Liscana (IDL) [].
2.4. Statistics
All data are presented as mean ± standard deviation. Data analysis was performed in a blinded fashion with respect to genotypes. GraphPad Prism was used for statistical inference and plotting (GraphPad Software, San Diego, CA, USA). To test for group differences, student’s t-test, Kruskal–Wallis One Way ANOVA on Ranks, or Chi-square test (dichotomous variables) was used. A p < 0.05 indicates significant statistical difference between groups.
3. Results
3.1. AF Inducibility in a Murine Model
In order to define the role of Gq-mediated signaling in AF in vivo, we used a transgenic mouse line with a tamoxifen-inducible, cardiomyocyte-specific Gαq/Gα11-deficiency (Gq-KO) []. Gq-KO and control mice underwent intracardiac electrophysiological studies using a 2F Octapolar catheter inserted via the right jugular vein. The inducibility of AF was determined before and after carbachol-induced vagal activation using a standardized protocol of programmed electrical stimulation []. Baseline electrophysiological parameters, including heart rate, sinus node recovery time, and atrial as well as AV nodal effective refractory periods, were comparable in Gq/G11-KO vs. control mice, with no significant differences between the two groups before or after carbachol-induced vagal activation (Figure 1). While AF could be induced in four out of 10 control mice, it was not inducible in any of the 11 Gq/G11-KO mice before carbachol administration (Figure 2A,B). Two minutes after vagal stimulation with 50 ng/g carbachol (i.p.), which resulted in a heart rate decrease of 15–20%, atrial pacing-induced AF in 8 out of 10 control mice (80%) but only in 3 out of 11 Gq/G11-KO mice (27%) (Figure 2C). Moreover, the mean duration of AF episodes was significantly shorter in Gq/G11-KO (23 ± 16 s) than in control mice (89 ± 14 s).
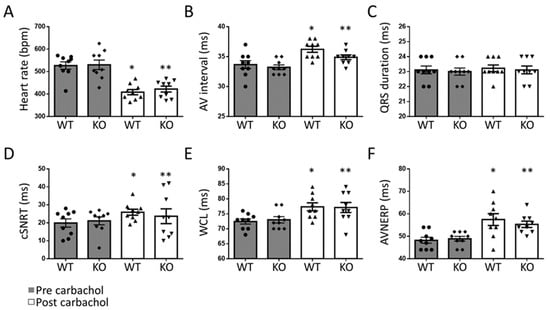
Figure 1.
Electrophysiological properties in Gq-KO (n = 11) and control mice (n = 10) before and after vagal stimulation with carbachol. Heart rate (A), atrioventricular (AV) interval (B), QRS duration (C), corrected sinus node recovery time (cSNRT, D), Wenckebach cycle length (WCL, E), and effective refractory period of the AV-node (AVNERP, F). * p < 0.05 vs. WT pre carbachol. ** p < 0.05 vs. KO pre carbachol.
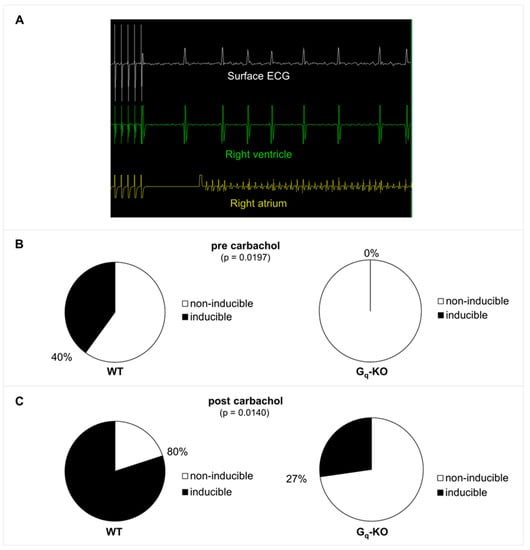
Figure 2.
(A) Example of AF induction by programmed stimulation in a wildtype control mouse before carbachol injection. There is a short blanking period in the atrial channel following pacing. (B) AF inducibility in Gq-KO (n = 11) versus control mice (n = 10) before application of carbachol. (C) AF inducibility 2 min after vagal stimulation with carbachol (intraperitoneal injection of 50 ng/g carbachol).
3.2. Baseline Characteristics of Excitation–Contraction Coupling in a Murine Model
We studied excitation–contraction coupling in WT and Gq-KO mice to elucidate further the role of the Gq pathway for pro-arrhythmogenic Ca2+ release. During electrical field stimulation, cytosolic Ca2+ transient amplitudes were unchanged upon Gq knockout (F/F0; 3.7 ± 0.2 vs. 3.3 ± 0.2 a.u. in Gq-KO, n.s.). Diastolic Ca2+ removal measured by assessment of the time-constant TAU (monoexponential fit of the Ca2+ decay phase) was also unaltered (98 ± 7 vs. 111 ± 8 ms in Gq-KO, n.s.). However, in Gq-KO, time to 50% of maximal Ca2+ release was significantly shortened (26 ± 2 vs. 22 ± 1 ms in Gq-KO, p < 0.05). In addition, nuclear Ca2+ release, as assessed by peak F/F0 within the nuclear compartment, was significantly reduced in atrial cardiomyocytes from Gq-KO animals (F/F0; 2.5 ± 0.2 vs. 2.0 ± 0.1, p < 0.05; Figure 3 and Supplementary Figure S1).
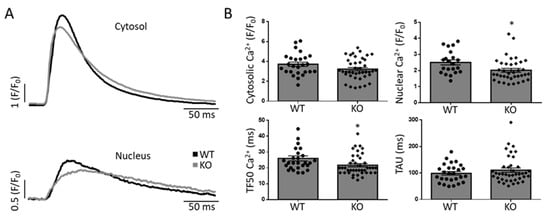
Figure 3.
Properties of Ca2+ signaling during excitation–contraction coupling in a murine model. Example of cytosolic and nuclear Ca2+ transients during field-stimulation (A) and quantification (B) of maximal cytosolic and nuclear Ca2+ release, time to 50% of maximal cytosolic Ca2+ release (TF50) and the time constant of Ca2+ decay/removal (TAU). * p < 0.05 vs. WT. Each data point represents an independent cell preparation and experiment (Cytosolic: WT n = 26, KO n = 41; nuclear: WT n = 21, KO n = 37; TF50: WT n = 27, KO n = 41; TAU: WT n = 27, KO n = 40). The total number of animals per group was n = 5 (see Supplementary Figure S1 for per-animal analyses).
3.3. Cellular and Subcellular Pro-Arrhythmogenic Ca2+ Release
Next, we tested the hypothesis of altered arrhythmogenic Ca2+ release at a (sub-) cellular level in Gq-KO. To obtain subcellular Ca2+ release properties, we quantified individual spontaneous Ca2+ spark characteristics in WT and Gq-KO (Figure 4, Supplementary Figure S1): Ca2+ sparks from Gq-KO animals were of equal amplitude and width, yet significantly shorter and with a decreased time to peak Ca2+ release (Figure 4B). This finding becomes even more apparent using histogram analysis of the Ca2+ spark full duration at half maximum and time to peak (Figure 4C). Overall, Ca2+ spark frequency did not differ significantly (3.8 ± 0.6 vs. 2.7 ± 0.4 in Gq-KO, p = 0.1); however, when corrected for SR Ca2+ content, Ca2+ sparks occurred less often in Gq-KO at a given Ca2+ content as compared to WT. In addition, with increasing SR Ca2+ content, the observed increase of Ca2+ spark frequency was less pronounced in Gq-KO as compared to WT (Figure 4D and Supplementary Figure S2).
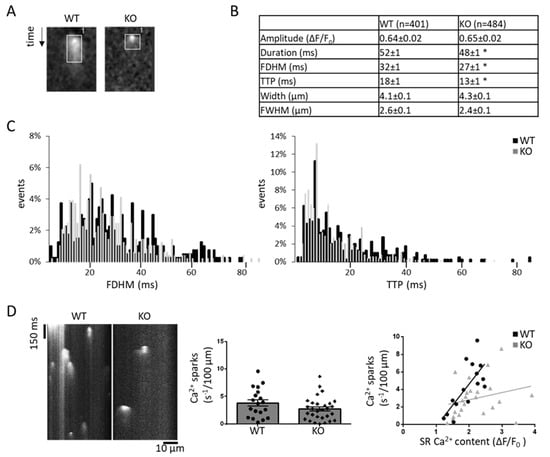
Figure 4.
Subcellular Ca2+ spark properties in a murine model. Example and quantification of Ca2+ sparks in WT and Gq-KO (A,B) as well as the distribution (C) of full duration at half maximum (FDHM) and time to peak (TTP) in all cells. Example for Ca2+ sparks in WT (n = 19) and Gq-KO (n = 29) and their respective overall frequency without and with correlation to the sarcoplasmic reticulum Ca2+ content, respectively, (WT n = 15, KO n = 23) (D). * p < 0.05 vs. WT. Each data point represents an independent cell preparation and experiment. The total number of animals per group was n = 5 (see Supplementary Figure S1 for per-animal analyses).
In line with this notion, we finally assessed cellular pro-arrhythmogenic Ca2+ wave activity: Ca2+ wave frequency was significantly reduced in Gq-KO compared to WT. Of note, Gq-KO also significantly altered Ca2+ wave propagation velocity in longitudinal line scans (Figure 5 and Supplementary Figure S1).
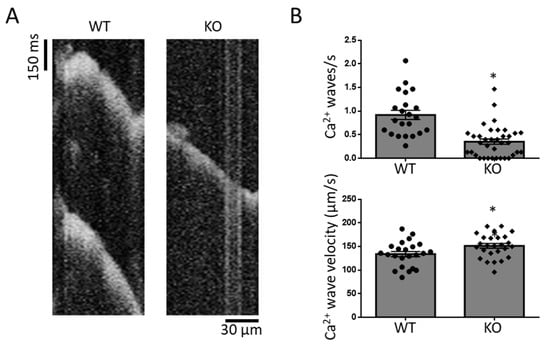
Figure 5.
Arrhythmogenic Ca2+ waves in WT and Gq-KO mice (A). Ca2+ wave frequency and propagation velocity (B). * p < 0.05 vs. WT. Each data point represents an independent cell preparation and experiment (Ca2+ wave frequency: WT n = 22, KO n = 36; Ca2+ wave velocity: WT n = 22, KO n = 26). The total number of animals per group was n = 5 (see Supplementary Figure S1 for per-animal analyses).
4. Discussion
Here we elucidate the central role of arrhythmogenic Gq-mediated signaling in the pathomechanism of AF. Cardiomyocyte-specific inactivation of Gq significantly reduced AF inducibility in a murine AF model. Our in vitro data in left atrial cardiomyocytes from Gq-KO mice indicate fewer spontaneous Ca2+ waves and altered Ca2+ spark properties as a potential mechanism of action.
4.1. Targeting Arrhythmogenic Gq-Signaling with Gq-Coupled Receptor Antagonists
Gq-signaling via IP3R is initiated by Gq-coupled receptors. Arrhythmogenic effects have been consistently demonstrated for the predominant cardiac Gq-coupled receptors, namely the angiotensin II receptor type 1 (AT1 receptor), the endothelin-1 receptor A (ETA receptor), the M3 muscarinic acetylcholine receptor (M3 receptor), thrombin (PAR) receptors and the alpha-1 adrenergic receptor []. In particular, abundant data implicate AT1 receptors in the pathogenesis of AF, and their inhibition has been shown to protect from AF in numerous animal models [].
While arrhythmogenic effects have in part been attributed to Gq-mediated profibrotic and proinflammatory signaling [,], direct proarrhythmogenic effects of AT1- and ETA-receptors have been increasingly appreciated in recent years [,]. In this regard, both angiotensin II-receptor type 1 and ET-1 receptors have been shown to enhance ectopic activity by promoting Ca2+ leak and delayed after-depolarisations [,,].
However, while indirect evidence from numerous clinical trials indicated that chronic inhibition of AT1 receptor signaling significantly reduces the incidence of AF, large randomized trials failed to demonstrate the beneficial effects of AT1 antagonists on AF [,,]. Against this background, it has to be considered that conventional AT1 antagonists do not selectively inhibit Gq-mediated signaling but equally block all downstream signaling pathways, some of which may even have beneficial effects. Thus, selective inhibition of Gq-mediated signaling may be desirable. This could be accomplished by biased ligands that act as Gq-selective antagonists. In fact, we have recently identified biased AT1-ligands that selectively inhibit Gq-mediated signaling (unpublished data). This angiotensin analog (TRV027) has proven to be well-tolerated and safe in phase II clinical trials in the context of heart failure. It may thus qualify as a suitable candidate for a Gq-targeting AF therapy [].
4.2. Gq-Dependent Mechanoelectrical Feedback
While many humoral stimuli can activate Gq-signaling through G-protein-coupled receptors, we have recently shown that this pathway is also mechanosensitive []. Mechanical stretch is a well-established determinant of atrial size and function, and immediate as well as chronic responses to mechanical forces can promote arrhythmia []. However, the molecular mechanisms that link mechanical forces to arrhythmogenesis are incompletely understood. The recent discovery of the mechanosensitive non-selective cation channel Piezo1 was a breakthrough in the field of mechanotransduction []. However, even though Piezo channels are expressed in the heart and have been implicated in cardiac arrhythmia, evidence of their cardiac function is still sparse.
Interestingly, we recently demonstrated that Piezo1 and Gq form a functional mechanosignaling complex in endothelial cells that may also be operative in cardiomyocytes []. This complex regulates IP3R-mediated Ca2+-signaling as well as NFκB-mediated proinflammatory signaling in response to mechanical forces—both key processes in the pathogenesis of AF. Thus, it is intriguing to speculate that Gq-mediated mechanotransduction is also involved in the arrhythmogenic mechanoelectrical feedback in the context of AF.
4.3. Gq-Signaling and Vagally-Dependent Atrial Fibrillation
Vagotonic conditions are known to promote AF, and in some patients, AF episodes are clearly vagally dependent [,]. Acetylcholine released by vagal nerve endings has been shown to stimulate Gi-coupled M2 muscarinergic receptors that activate G-protein- gated K+ channels, thereby reducing atrial action potential duration and increasing susceptibility to early after-depolarisations as well as reentrant mechanisms []. However, while arrhythmogenic effects have been largely attributed to Gi-mediated signaling downstream of M2 muscarinic receptors, Gq-coupled M3 muscarinic receptors are also expressed in the atria and appear to mediate arrhythmogenic effects of vagal activation to some extent [,]. These arrhythmogenic M3 effects seem to involve DADs generated by abnormal Ca2+ events. Thus, in light of the data presented here, the relative contribution of Gi-coupled M2 vs. Gq-coupled M3 receptors to vagally dependent AF may have to be reconsidered.
4.4. The Role of the Gq Pathway for SR Ca2+ Leak and Pro-Arrhythmogenic Cellular Conditions
IP3R-mediated Ca2+ release has been shown to also facilitate SR Ca2+ release via sensitization of nearby RyR clusters []. Atrial cardiomyocytes from Gq-KO animals showed decreased spontaneous SR Ca2+ release in support of this notion. Interestingly, cytosolic Ca2+ release during excitation–contraction coupling, i.e., Ca2+ transient amplitude, was not significantly affected by Gq knockout, indicating unaltered baseline Ca2+ signaling. Of note, spontaneous Ca2+ release events were reported to be increased in human atrial cardiomyocytes during chronic AF, and enhanced SR Ca2+ leak has been associated with DADs in this setting []. Normalizing SR Ca2+ leak, e.g., through genetic inhibition of Ca2+/calmodulin-dependent protein kinase II-mediated (CaMKII) RyR2-S2814 phosphorylation, was shown to delay the development of spontaneous atrial ectopy and fully prevent AF in mice []. Gq-mediated signaling altering SR Ca2+ release might represent another piece of the puzzle of IP3R-dependent SR Ca2+ leak causally linked to the development of AF.
As the use of all currently approved antiarrhythmic drugs is limited by potentially life-threatening ventricular pro-arrhythmic effects, from a translational perspective, it is intriguing that IP3R expression in atrial myocytes is 6- to 10-fold higher than in ventricular myocytes [,], and that IP3R-mediated electrophysiological effects on Ca2+ homeostasis are absent in ventricular myocytes [].
We also report a decrease of nuclear Ca2+ transient amplitudes upon Gq knockout, further underscoring the notion of abundant IP3R expression in the nuclear envelope []. These findings have important ramifications in the setting of AF: Only recently has AF been shown to increase atrial-cardiomyocyte nucleoplasmic Ca2+ by IP3R-upregulation, leading to enhanced IP3R-CaMKII-HDAC4 signaling and L-type calcium current downregulation []. Altered nuclear Ca2+, mediated via the Gq signaling cascade, might, therefore, directly affect gene regulation important for Ca2+ release and SR Ca2+ leak in the setting of AF.
4.5. Altered Calcium Handling as an Arrhythmogenic Substrate
The impact of increased calcium release from the sarcoplasmic reticulum on AF inducibility in vivo may be counterintuitive, as it has primarily been regarded as a mechanism of triggered activity-inducing AF rather than an arrhythmogenic substrate sustaining AF. However, our findings are in line with a number of previous studies in which diastolic calcium leak from the sarcoplasmic reticulum was not associated with triggered activity resulting in spontaneous AF events but with an increased AF inducibility by programmed stimulation in mice [,]. Taken together, these data indicate that altered calcium release can create an arrhythmogenic substrate favoring AF initiation and maintenance.
5. Conclusions
Our combined in vitro and in vivo studies in mice with cardiomyocyte-specific Gq-deficiency demonstrate that Gq-mediated signal transduction promotes arrhythmogenic Ca2+-release and AF in mice. These data suggest that Gq-signaling likely mediates arrhythmogenic effects of angiotensin II, vagal stimulation, mechanical stress, and other stimuli known to promote AF. Thus, targeting the Gq-pathway, ideally using Gq-selective biased receptor ligands, may be a promising approach for the treatment and prevention of AF. Importantly, the atrial-selective expression of the Gq-effector IP3R confers atrial selectivity mitigating the risk of life-threatening ventricular pro-arrhythmic effects.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines11020526/s1, Figure S1: Per-animal analyses of Ca2+ signaling. Figure S2: SR Ca2+ content in WT and KO mice as obtained with caffeine (top).
Author Contributions
Conceptualization, T.F.A. and F.H.; methodology, T.F.A. and F.H.; formal analysis F.H., A.P., T.F.A.; investigation, F.H., A.P., T.F.A.; resources, V.S., B.P., K.S., N.W., T.F.A.; writing—original draft preparation, T.F.A. and F.H.; writing—review and editing, all authors; supervision, T.F.A. and F.H.; project administration, T.F.A. and F.H.; funding acquisition, T.F.A. and F.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the German Research Foundation (DFG) [AL 1795/3-1 to T.F.A.; HO 5647/4-1 to F.H. and 437531118—SFB 1470 to N.W. and F.H.].
Institutional Review Board Statement
All animal experiments were approved by the responsible federal authority (LAGeSo Berlin, approval TVA G0006/18) and performed conforming to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Reporting in the manuscript follows the recommendations in the ARRIVE guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data underlying this article will be shared upon reasonable request to the corresponding author.
Acknowledgments
We wish to thank Andrea Weller for her expert technical assistance.
Conflicts of Interest
The authors declare no competing interests.
References
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2020, 42, 373–498. [Google Scholar] [CrossRef]
- Guerra, P.G.; Talajic, M.; Roy, D.; Dubuc, M.; Thibault, B.; Nattel, S. Is there a future for antiarrhythmic drug therapy? Drugs 1998, 56, 767–781. [Google Scholar] [CrossRef]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on Atrial cardiomyopathies: Definition, characterisation, and clinical implication. J. Arrhythm. 2016, 32, 247–278. [Google Scholar] [CrossRef]
- Tinker, A.; Finlay, M.; Nobles, M.; Opel, A. The contribution of pathways initiated via the G G-protein family to atrial fibrillation. Pharmacol. Res. 2016, 105, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.R.; Bode, F. Mechano-electrical feedback underlying arrhythmias: The atrial fibrillation case. Prog. Biophys. Mol. Biol. 2003, 82, 163–174. [Google Scholar] [CrossRef]
- Albarran-Juarez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef]
- Pabel, S.; Ahmad, S.; Tirilomis, P.; Stehle, T.; Mustroph, J.; Knierim, M.; Dybkova, N.; Bengel, P.; Holzamer, A.; Hilker, M.; et al. Inhibition of Na(V)1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res. Cardiol. 2020, 115, 20. [Google Scholar] [CrossRef]
- Li, X.; Zima, A.V.; Sheikh, F.; Blatter, L.A.; Chen, J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ. Res. 2005, 96, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Ohkusa, T.; Nao, T.; Ueyama, T.; Yano, M.; Kobayashi, S.; Hamano, K.; Esato, K.; Matsuzaki, M. Up-regulation of inositol 1, 4, 5-trisphosphate receptor expression in atrial tissue in patients with chronic atrial fibrillation. J. Cardiol. 2002, 39, 57–58. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. J. Physiol. 2004, 555, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, L.; Bootman, M.D.; Laine, M.; Berridge, M.J.; Thuring, J.; Holmes, A.; Li, W.H.; Lipp, P. The role of inositol 1,4,5-trisphosphate receptors in Ca(2+) signalling and the generation of arrhythmias in rat atrial myocytes. J. Physiol. 2002, 541, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Rütten, H.; Zywietz, A.; Gehring, D.; Wilkie, T.M.; Chen, J.; Chien, K.R.; Offermanns, S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 2001, 7, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Takefuji, M.; Wirth, A.; Lukasova, M.; Takefuji, S.; Boettger, T.; Braun, T.; Althoff, T.; Offermanns, S.; Wettschureck, N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation 2012, 126, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Maguire, C.T.; Kovoor, P.; Hammer, P.E.; Gehrmann, J.; Triedman, J.K.; Berul, C.I. Induction of atrial tachycardia and fibrillation in the mouse heart. Cardiovasc. Res. 2001, 50, 463–473. [Google Scholar] [CrossRef]
- Hohendanner, F.; Ljubojevic, S.; MacQuaide, N.; Sacherer, M.; Sedej, S.; Biesmans, L.; Wakula, P.; Platzer, D.; Sokolow, S.; Herchuelz, A.; et al. Intracellular dyssynchrony of diastolic cytosolic [Ca(2)(+)] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ. Res. 2013, 113, 527–538. [Google Scholar] [CrossRef]
- Dimai, S.; Semmler, L.; Prabhu, A.; Stachelscheid, H.; Huettemeister, J.; Klaucke, S.C.; Lacour, P.; Blaschke, F.; Kruse, J.; Parwani, A.; et al. COVID19-associated cardiomyocyte dysfunction, arrhythmias and the effect of Canakinumab. PLoS ONE 2021, 16, e0255976. [Google Scholar] [CrossRef]
- Hohendanner, F.; Maxwell, J.T.; Blatter, L.A. Cytosolic and nuclear calcium signaling in atrial myocytes: IP3-mediated calcium release and the role of mitochondria. Channels 2015, 9, 129–138. [Google Scholar] [CrossRef]
- Picht, E.; Zima, A.V.; Blatter, L.A.; Bers, D.M. SparkMaster: Automated calcium spark analysis with ImageJ. Am. J. Physiol. Cell Physiol. 2007, 293, C1073–C1081. [Google Scholar] [CrossRef]
- Hohendanner, F.; Walther, S.; Maxwell, J.T.; Kettlewell, S.; Awad, S.; Smith, G.L.; Lonchyna, V.A.; Blatter, L.A. Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J. Physiol. 2015, 593, 1459–1477. [Google Scholar] [CrossRef]
- Verheule, S.; Sato, T.; Everett, T.T.; Engle, S.K.; Otten, D.; Rubart-von der Lohe, M.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef]
- Nakashima, H.; Kumagai, K.; Urata, H.; Gondo, N.; Ideishi, M.; Arakawa, K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation 2000, 101, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.C.; Kleeschulte, S.; Stolte, S.; Leineweber, K.; Kamler, M.; Bode, J.; Li, N.; Dobrev, D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res. Cardiol. 2020, 115, 10. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, Y.C.; Tai, C.T.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Angiotensin II and angiotensin II receptor blocker modulate the arrhythmogenic activity of pulmonary veins. Br. J. Pharmacol. 2006, 147, 12–22. [Google Scholar] [CrossRef] [PubMed]
- von Lewinski, D.; Kockskamper, J.; Rubertus, S.U.; Zhu, D.; Schmitto, J.D.; Schondube, F.A.; Hasenfuss, G.; Pieske, B. Direct pro-arrhythmogenic effects of angiotensin II can be suppressed by AT1 receptor blockade in human atrial myocardium. Eur. J. Heart Fail 2008, 10, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Schon, N.; Kirchhof, P.; Breithardt, G.; Fetsch, T.; Hausler, K.G.; Klein, H.U.; Steinbeck, G.; Wegscheider, K.; Meinertz, T. Angiotensin II-antagonist in paroxysmal atrial fibrillation (ANTIPAF) trial. Circ. Arrhythm. Electrophysiol. 2012, 5, 43–51. [Google Scholar] [CrossRef]
- Investigators, G.-A.; Disertori, M.; Latini, R.; Barlera, S.; Franzosi, M.G.; Staszewsky, L.; Maggioni, A.P.; Lucci, D.; Di Pasquale, G.; Tognoni, G. Valsartan for prevention of recurrent atrial fibrillation. N. Engl. J. Med. 2009, 360, 1606–1617. [Google Scholar] [CrossRef]
- Schneider, M.P.; Hua, T.A.; Bohm, M.; Wachtell, K.; Kjeldsen, S.E.; Schmieder, R.E. Prevention of atrial fibrillation by Renin-Angiotensin system inhibition a meta-analysis. J. Am. Coll. Cardiol. 2010, 55, 2299–2307. [Google Scholar] [CrossRef]
- Pang, P.S.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: A randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 2017, 38, 2364–2373. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sharifov, O.F.; Fedorov, V.V.; Beloshapko, G.G.; Glukhov, A.V.; Yushmanova, A.V.; Rosenshtraukh, L.V. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J. Am. Coll. Cardiol. 2004, 43, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, M.; Zimmermann, M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002, 105, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Nguyen, B.L.; Tan, A.Y.; Chang, P.C.; Lee, H.L.; Lin, F.C.; Yeh, S.J.; Fishbein, M.C.; Lin, S.F.; Wu, D.; et al. Intracellular calcium dynamics and acetylcholine-induced triggered activity in the pulmonary veins of dogs with pacing-induced heart failure. Heart Rhythm 2008, 5, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, J.M.; Chidiac, P.; Jones, D.L. Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2-deficient mouse. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H554–H561. [Google Scholar] [CrossRef]
- Kitazawa, T.; Asakawa, K.; Nakamura, T.; Teraoka, H.; Unno, T.; Komori, S.; Yamada, M.; Wess, J. M3 muscarinic receptors mediate positive inotropic responses in mouse atria: A study with muscarinic receptor knockout mice. J. Pharmacol. Exp. Ther. 2009, 330, 487–493. [Google Scholar] [CrossRef]
- Li, N.; Chiang, D.Y.; Wang, S.; Wang, Q.; Sun, L.; Voigt, N.; Respress, J.L.; Ather, S.; Skapura, D.G.; Jordan, V.K.; et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 2014, 129, 1276–1285. [Google Scholar] [CrossRef]
- Lipp, P.; Laine, M.; Tovey, S.C.; Burrell, K.M.; Berridge, M.J.; Li, W.; Bootman, M.D. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr. Biol. 2000, 10, 939–942. [Google Scholar] [CrossRef]
- Qi, X.Y.; Vahdati Hassani, F.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2021, 128, 619–635. [Google Scholar] [CrossRef]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Muller, F.U.; Schmitz, W.; et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Investig. 2009, 119, 1940–1951. [Google Scholar] [CrossRef]
- Sood, S.; Chelu, M.G.; van Oort, R.J.; Skapura, D.; Santonastasi, M.; Dobrev, D.; Wehrens, X.H. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 2008, 5, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).