

Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research
 , , , ,
, , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Animals
2.3. Cells
2.4. Drugs
2.5. Tumour Cell Implantation
2.6. Tumour Growth Assessment with Bioluminescence
2.7. Histological Analysis
2.8. Data and Statistical Analysis
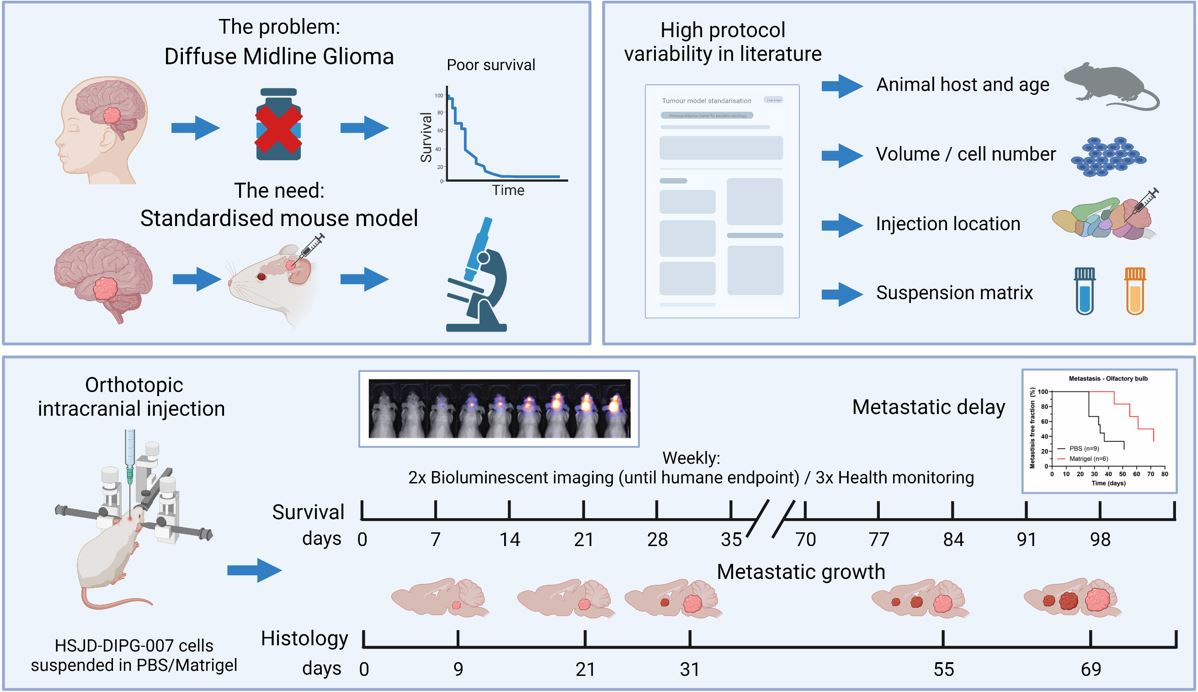
3. Results
3.1. HSJD-DIPG-007 PDX Model in the Literature
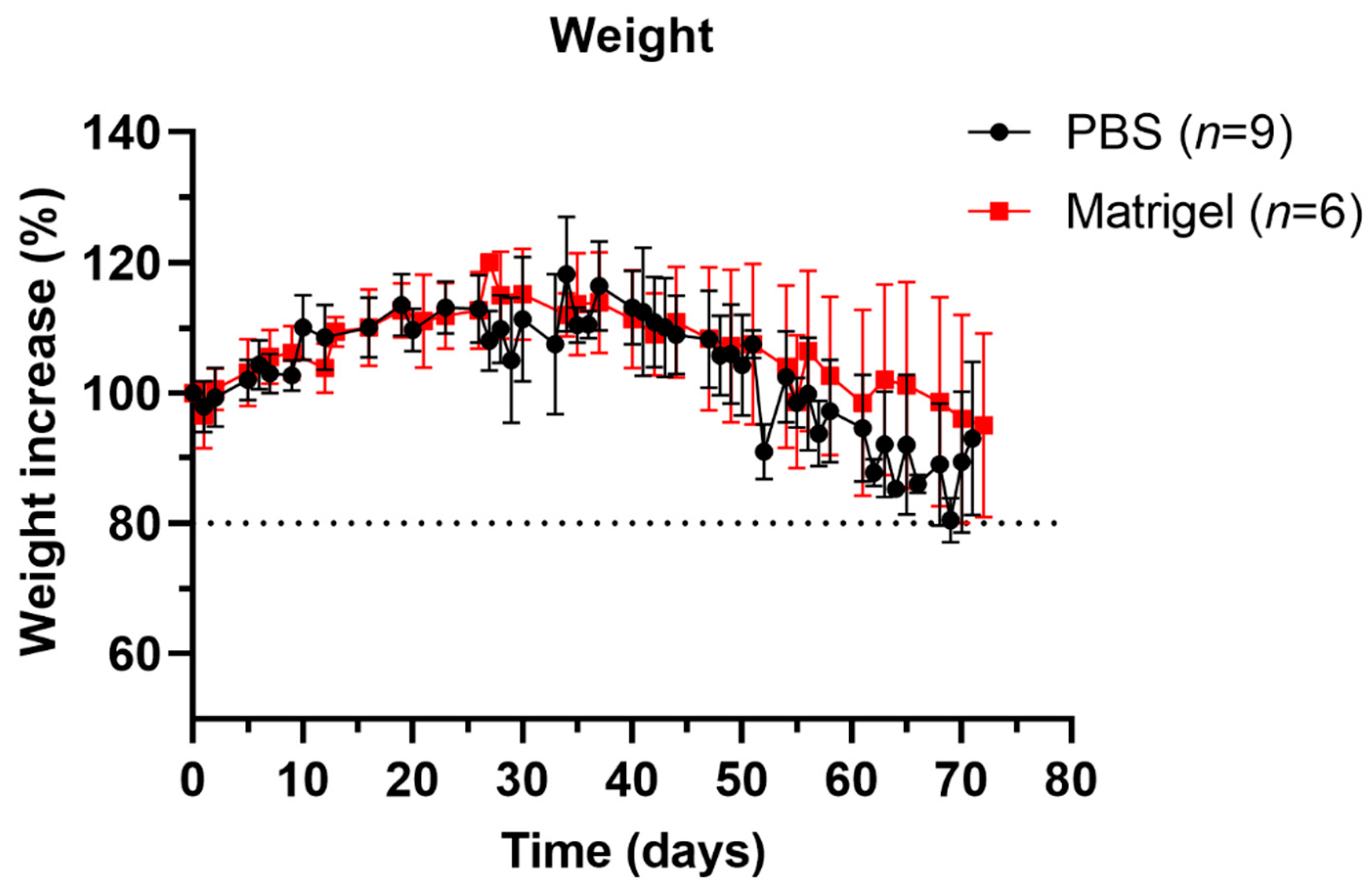
3.2. Well-Being and Weight Profiles upon Implantation Procedure
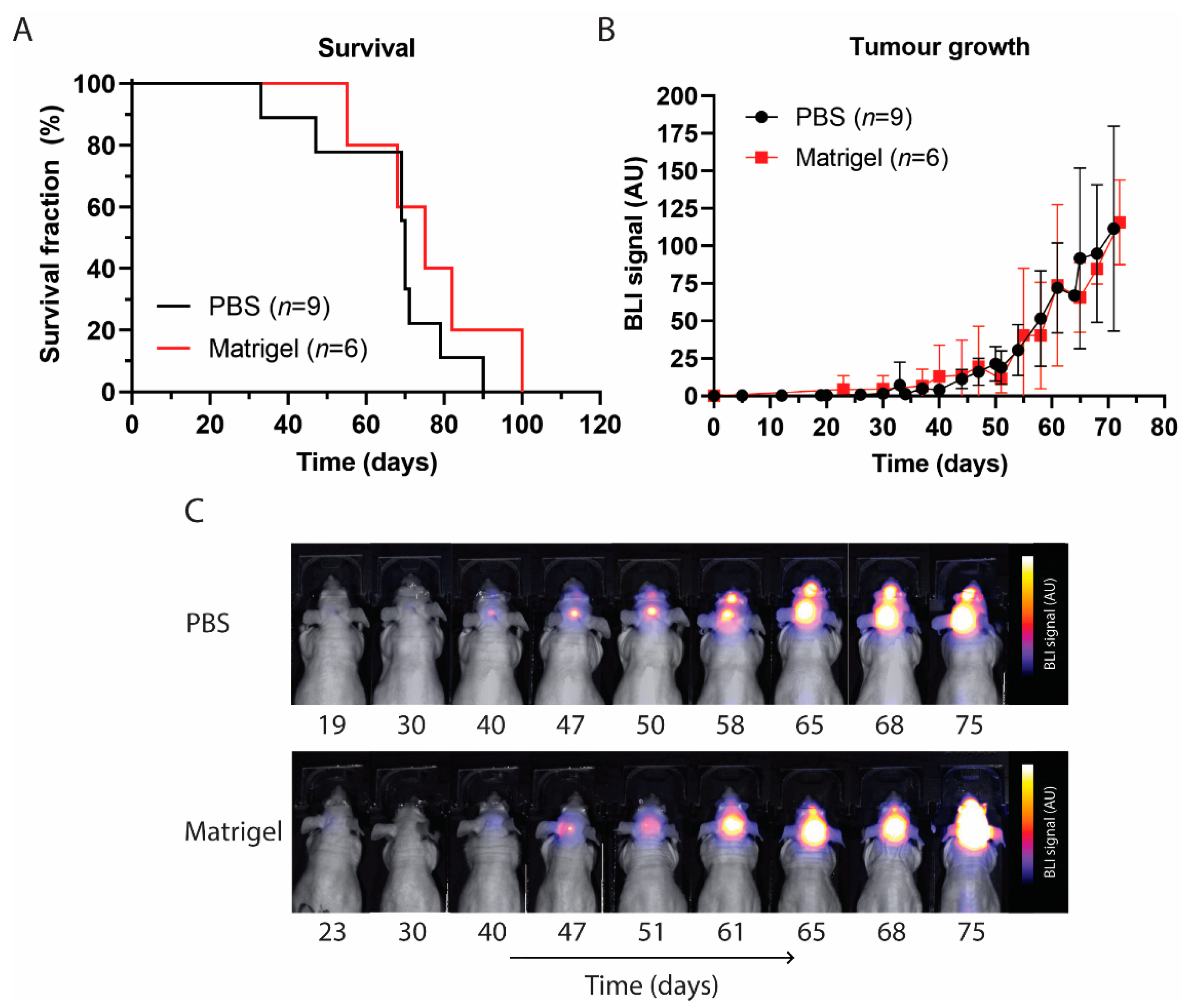
3.3. Survival and Tumour Growth Using PBS or Matrigel as Suspension Matrices
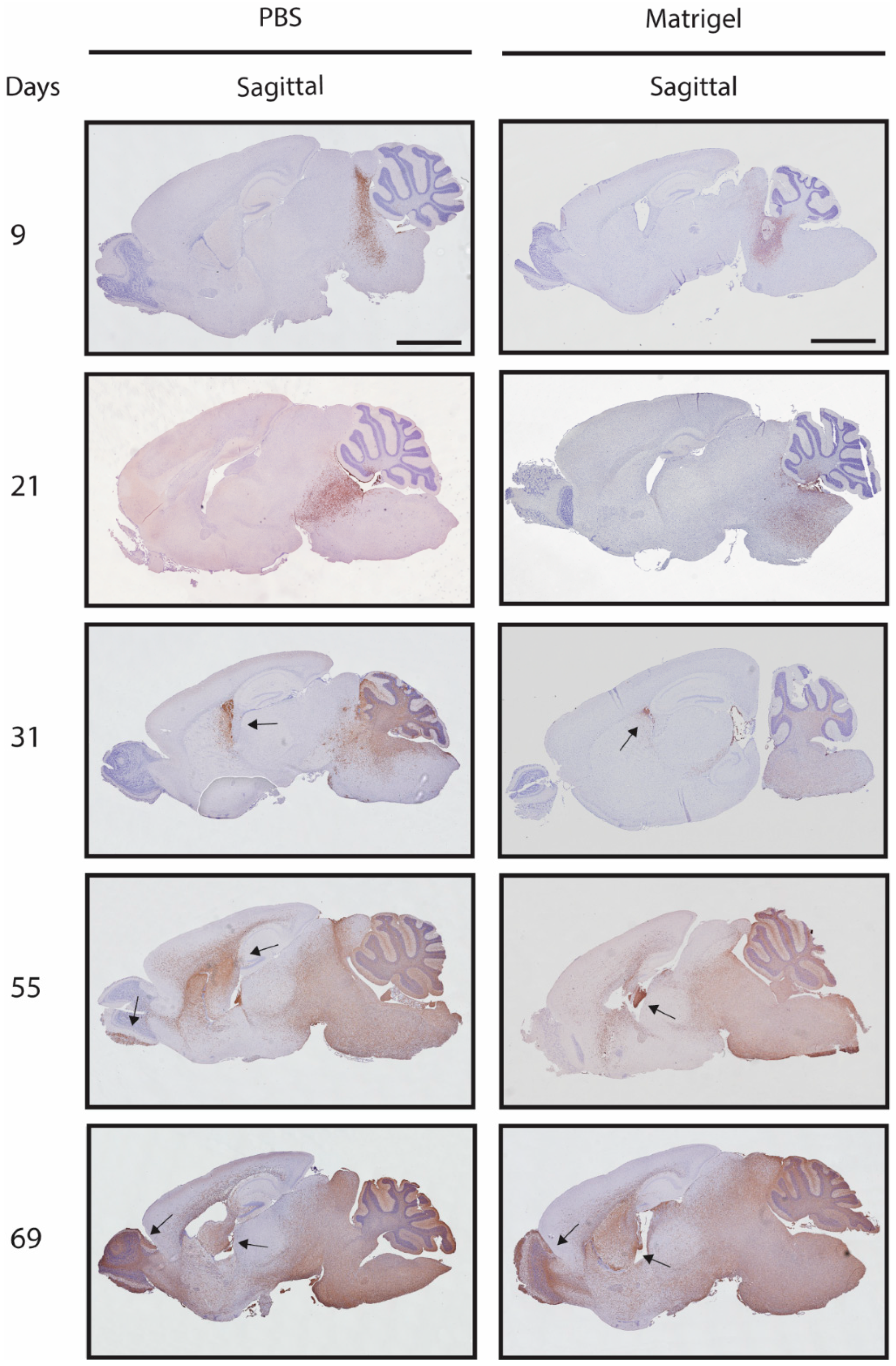
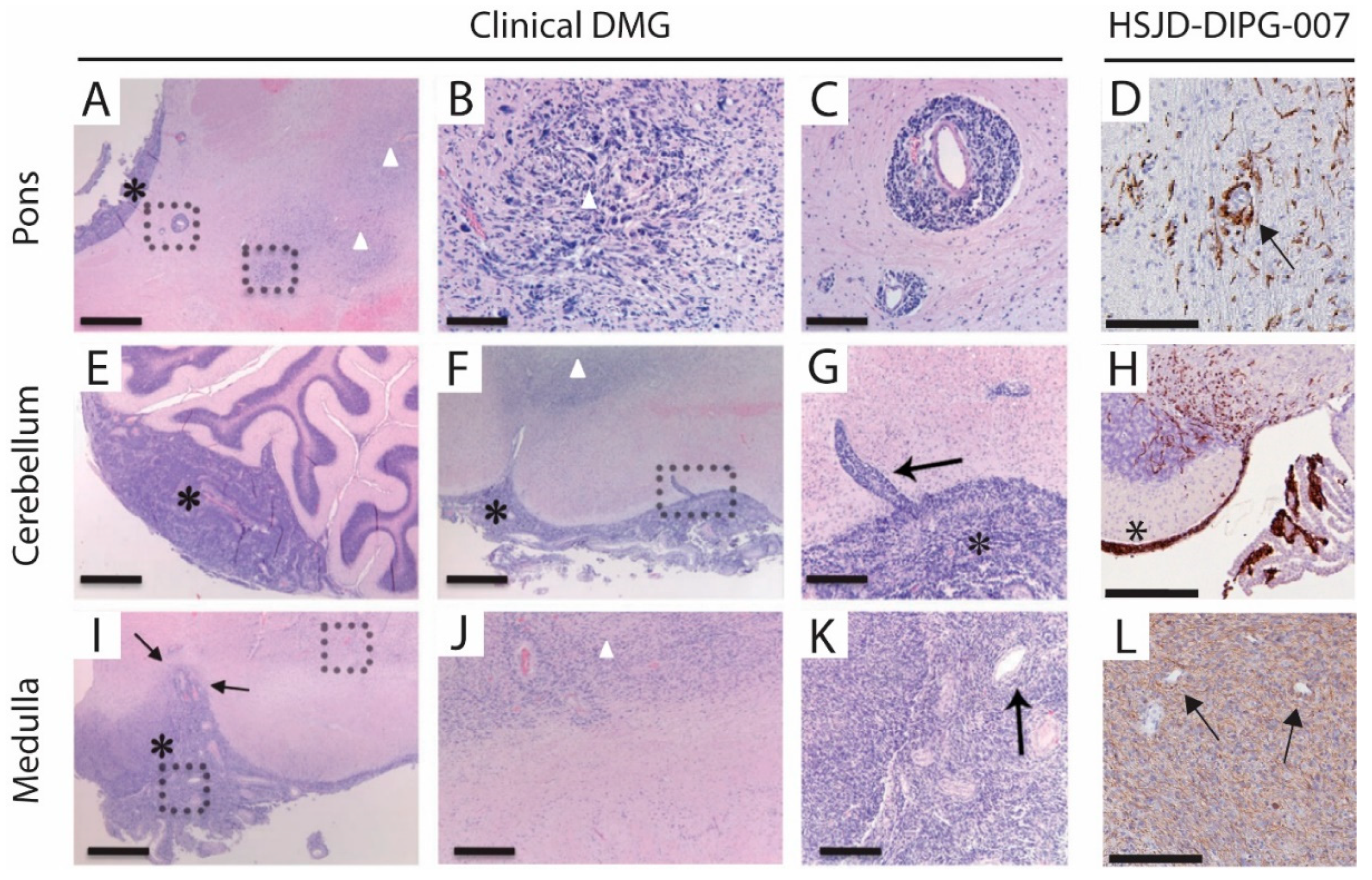
3.4. Metastases Occurrence in Olfactory Bulb and Spinal Cord
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M Mutation in Histone H3.3 Defines Clinically and Biologically Distinct Subgroups of Pediatric Diffuse Intrinsic Pontine Gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The Histone H3.3K27M Mutation in Pediatric Glioma Reprograms H3K27 Methylation and Gene Expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 Wild-Type DIPG/DMG Overexpressing EZHIP Extend the Spectrum Diffuse Midline Gliomas with PRC2 Inhibition beyond H3-K27M Mutation. Acta Neuropathol. 2020, 139, 1109–1113. [Google Scholar] [CrossRef]
- Xu, C.; Liu, H.; Pirozzi, C.J.; Chen, L.H.; Greer, P.K.; Diplas, B.H.; Zhang, L.; Waitkus, M.S.; He, Y.; Yan, H. TP53 Wild-Type/PPM1D Mutant Diffuse Intrinsic Pontine Gliomas Are Sensitive to a MDM2 Antagonist. Acta Neuropathol. Commun. 2021, 9, 178. [Google Scholar] [CrossRef]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor Alpha and Poly (ADP-Ribose) Polymerase as Potential Therapeutic Targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; da Silva, N.S.; et al. Genome-Wide Analyses Identify Recurrent Amplifications of Receptor Tyrosine Kinases and Cell-Cycle Regulatory Genes in Diffuse Intrinsic Pontine Glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic Analysis of Diffuse Intrinsic Pontine Gliomas Identifies Three Molecular Subgroups and Recurrent Activating ACVR1 Mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.H.; Wan, H.; Yang, R.; Wang, Z.; Feng, J.; Yang, S.; Jones, S.; Wang, S.; Zhou, W.; et al. Exome Sequencing Identifies Somatic Gain-of-Function PPM1D Mutations in Brainstem Gliomas. Nat. Genet. 2014, 46, 726–730. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report from the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef]
- Jansen, M.H.; Veldhuijzen van Zanten, S.E.; Sanchez Aliaga, E.; Heymans, M.W.; Warmuth-Metz, M.; Hargrave, D.; van der Hoeven, E.J.; Gidding, C.E.; de Bont, E.S.; Eshghi, O.S.; et al. Survival Prediction Model of Children with Diffuse Intrinsic Pontine Glioma Based on Clinical and Radiological Criteria. Neuro. Oncol. 2015, 17, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.C.; Lindquist, R.A.; Nguyen, T.; Sanai, N.; Barkovich, A.J.; Huang, E.J.; Rowitch, D.H.; Alvarez-Buylla, A. Postnatal Growth of the Human Pons: A Morphometric and Immunohistochemical Analysis. J. Comp. Neurol. 2015, 523, 449–462. [Google Scholar] [CrossRef]
- Fisher, P.G.; Breiter, S.N.; Carson, B.S.; Wharam, M.D.; Williams, J.A.; Weingart, J.D.; Foer, D.R.; Goldthwaite, P.T.; Tihan, T.; Burger, P.C. A Clinicopathologic Reappraisal of Brain Stem Tumor Classification. Identification of Pilocystic Astrocytoma and Fibrillary Astrocytoma as Distinct Entities. Cancer 2000, 89, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier Delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Warren, K.E. Beyond the Blood:Brain Barrier: The Importance of Central Nervous System (CNS) Pharmacokinetics for the Treatment of CNS Tumors, Including Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Gururangan, S.; McLaughlin, C.A.; Brashears, J.; Watral, M.A.; Provenzale, J.; Coleman, R.E.; Halperin, E.C.; Quinn, J.; Reardon, D.; Vredenburgh, J.; et al. Incidence and Patterns of Neuraxis Metastases in Children with Diffuse Pontine Glioma. J. Neurooncol. 2006, 77, 207–212. [Google Scholar] [CrossRef]
- Caretti, V.; Bugiani, M.; Freret, M.; Schellen, P.; Jansen, M.; van Vuurden, D.; Kaspers, G.; Fisher, P.G.; Hulleman, E.; Wesseling, P.; et al. Subventricular Spread of Diffuse Intrinsic Pontine Glioma. Acta Neuropathol. 2014, 128, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse Brainstem Glioma in Children: Critical Review of Clinical Trials. Lancet Oncol. 2006, 7, 241–248. [Google Scholar] [CrossRef]
- Veringa, S.J.E.; Biesmans, D.; van Vuurden, D.G.; Jansen, M.H.A.; Wedekind, L.E.; Horsman, I.; Wesseling, P.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J.L.; et al. In Vitro Drug Response and Efflux Transporters Associated with Drug Resistance in Pediatric High Grade Glioma and Diffuse Intrinsic Pontine Glioma. PLoS ONE 2013, 8, e61512. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally Defined Therapeutic Targets in Diffuse Intrinsic Pontine Glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the Treatment of Pediatric High-Grade Gliomas. Neuro. Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.L.; Noske, D.P.; et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2013, 12, 141–150. [Google Scholar] [CrossRef]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, P.; Zhang, X.; Mania-Farnell, B.; Xi, G.; Wan, F. Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers 2021, 13, 1114. [Google Scholar] [CrossRef]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of Human Embryonic Stem Cells to Model Pediatric Gliomas with H3.3K27M Histone Mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.B.; et al. Hedgehog-Responsive Candidate Cell of Origin for Diffuse Intrinsic Pontine Glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent Activating ACVR1 Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional Diversity and Cooperativity between Subclonal Populations of Pediatric Glioblastoma and Diffuse Intrinsic Pontine Glioma Cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef]
- Haumann, R.; Bianco, J.I.; Waranecki, P.M.; Gaillard, P.J.; Storm, G.; Ries, M.; van Vuurden, D.G.; Kaspers, G.J.L.; Hulleman, E. Imaged-Guided Focused Ultrasound in Combination with Various Formulations of Doxorubicin for the Treatment of Diffuse Intrinsic Pontine Glioma. Transl. Med. Commun. 2022, 7, 8. [Google Scholar] [CrossRef]
- Brust, V.; Schindler, P.M.; Lewejohann, L. Lifetime Development of Behavioural Phenotype in the House Mouse (Mus Musculus). Front. Zool. 2015, 12, S17. [Google Scholar] [CrossRef]
- Qi, L.; Kogiso, M.; Du, Y.; Zhang, H.; Braun, F.K.; Huang, Y.; Teo, W.-Y.; Lindsay, H.; Zhao, S.; Baxter, P.; et al. Impact of SCID Mouse Gender on Tumorigenicity, Xenograft Growth and Drug-Response in a Large Panel of Orthotopic PDX Models of Pediatric Brain Tumors. Cancer Lett. 2020, 493, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Kholosy, W.M.; Derieppe, M.; van den Ham, F.; Ober, K.; Su, Y.; Custers, L.; Schild, L.; van Zogchel, L.; Wellens, L.; Ariese, H.; et al. Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies. J. Pers. Med. 2021, 11, 869. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Meel, M.H.; de Gooijer, M.C.; Metselaar, D.S.; Sewing, A.C.P.; Zwaan, K.; Waranecki, P.; Breur, M.; Buil, L.C.M.; Lagerweij, T.; Wedekind, L.E.; et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020, 26, 3319–3332. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Smith, N.; Saunders, D.; Zalles, M.; Gulej, R.; Lerner, M.; Fung, K.-M.; Carcaboso, A.M.; Towner, R.A. OKlahoma Nitrone-007: Novel Treatment for Diffuse Intrinsic Pontine Glioma. J. Transl. Med. 2020, 18, 424. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.-J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349. [Google Scholar] [CrossRef]
- Chaves, C.; Declèves, X.; Taghi, M.; Menet, M.-C.; Lacombe, J.; Varlet, P.; Olaciregui, N.G.; Carcaboso, A.M.; Cisternino, S. Characterization of the Blood-Brain Barrier Integrity and the Brain Transport of SN-38 in an Orthotopic Xenograft Rat Model of Diffuse Intrinsic Pontine Glioma. Pharmaceutics 2020, 12, 399. [Google Scholar] [CrossRef]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; et al. Targeting Reduced Mitochondrial DNA Quantity as a Therapeutic Approach in Pediatric High-Grade Gliomas. Neuro Oncol. 2020, 22, 139–151. [Google Scholar] [CrossRef]
- Ehteda, A.; Simon, S.; Franshaw, L.; Giorgi, F.M.; Liu, J.; Joshi, S.; Rouaen, J.R.C.; Pang, C.N.I.; Pandher, R.; Mayoh, C.; et al. Dual Targeting of the Epigenome via FACT Complex and Histone Deacetylase Is a Potent Treatment Strategy for DIPG. Cell Rep. 2021, 35, 108994. [Google Scholar] [CrossRef] [PubMed]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal Stem Cells Successfully Deliver Oncolytic Virotherapy to Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2021, 27, 1766–1777. [Google Scholar] [CrossRef]
- Surowiec, R.K.; Ferris, S.F.; Apfelbaum, A.; Espinoza, C.; Mehta, R.K.; Monchamp, K.; Sirihorachai, V.R.; Bedi, K.; Ljungman, M.; Galban, S. Transcriptomic Analysis of Diffuse Intrinsic Pontine Glioma (DIPG) Identifies a Targetable ALDH-Positive Subset of Highly Tumorigenic Cancer Stem-like Cells. Mol. Cancer Res. 2021, 19, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Gamble, L.D.; Upton, D.H.; Ung, C.; Yu, D.M.T.; Ehteda, A.; Pandher, R.; Mayoh, C.; Hébert, S.; Jabado, N.; et al. Dual Targeting of Polyamine Synthesis and Uptake in Diffuse Intrinsic Pontine Gliomas. Nat. Commun. 2021, 12, 971. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.M.; Richardson, P.J.; Olaciregui, N.; Stankunaite, R.; Lavarino, C.; Molinari, V.; Corley, E.A.; Smith, D.P.; Ruddle, R.; Donovan, A.; et al. Repurposing Vandetanib plus Everolimus for the Treatment of ACVR1-Mutant Diffuse Intrinsic Pontine Glioma. Cancer Discov. 2022, 12, 416–431. [Google Scholar] [CrossRef]
- Jansen, M.H.A.; Lagerweij, T.; Sewing, A.C.P.; Vugts, D.J.; van Vuurden, D.G.; Molthoff, C.F.M.; Caretti, V.; Veringa, S.J.E.; Petersen, N.; Carcaboso, A.M.; et al. Bevacizumab Targeting Diffuse Intrinsic Pontine Glioma: Results of 89Zr-Bevacizumab PET Imaging in Brain Tumor Models. Mol. Cancer Ther. 2016, 15, 2166–2174. [Google Scholar] [CrossRef]
- Sewing, A.C.P.; Lagerweij, T.; van Vuurden, D.G.; Meel, M.H.; Veringa, S.J.E.; Carcaboso, A.M.; Gaillard, P.J.; Peter Vandertop, W.; Wesseling, P.; Noske, D.; et al. Preclinical Evaluation of Convection-Enhanced Delivery of Liposomal Doxorubicin to Treat Pediatric Diffuse Intrinsic Pontine Glioma and Thalamic High-Grade Glioma. J. Neurosurg. Pediatr. 2017, 19, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.; Brüning-Richardson, A.; Scott, K.J.; Thompson, J.; Kottke, T.; Morrison, E.; Ismail, A.; Carcaboso, A.M.; Rose, A.; Selby, P.; et al. Oncolytic Herpes Simplex Virus Inhibits Pediatric Brain Tumor Migration and Invasion. Mol. Ther. Oncolytics 2017, 5, 75–86. [Google Scholar] [CrossRef]
- Tsoli, M.; Shen, H.; Mayoh, C.; Franshaw, L.; Ehteda, A.; Upton, D.; Carvalho, D.; Vinci, M.; Meel, M.H.; van Vuurden, D.; et al. International Experience in the Development of Patient-Derived Xenograft Models of Diffuse Intrinsic Pontine Glioma. J. Neurooncol. 2019, 141, 253–263. [Google Scholar] [CrossRef]
- Akamandisa, M.P.; Nie, K.; Nahta, R.; Hambardzumyan, D.; Castellino, R.C. Inhibition of Mutant PPM1D Enhances DNA Damage Response and Growth Suppressive Effects of Ionizing Radiation in Diffuse Intrinsic Pontine Glioma. Neuro Oncol. 2019, 21, 786–799. [Google Scholar] [CrossRef]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 Inhibitors Display Beneficial Effects in Preclinical Models of ACVR1 Mutant Diffuse Intrinsic Pontine Glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Zondervan, I.; Meijer, D.H.; Idema, S.; Vos, W.; Hamans, B.; Bugiani, M.; Hulleman, E.; Wesseling, P.; Vandertop, W.P.; et al. Monitoring of Tumor Growth and Post-Irradiation Recurrence in a Diffuse Intrinsic Pontine Glioma Mouse Model. Brain Pathol. 2011, 21, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-J.; Lee, C.-L.; Wang, Q.; Zhou, Z.-W.; Yang, F.; Jin, C.; Fu, D.-L. Establishment of an Orthotopic Pancreatic Cancer Mouse Model: Cells Suspended and Injected in Matrigel. World J. Gastroenterol. 2014, 20, 9476–9485. [Google Scholar] [CrossRef]
- Kleinman, H.K.; McGarvey, M.L.; Liotta, L.A.; Robey, P.G.; Tryggvason, K.; Martin, G.R. Isolation and Characterization of Type IV Procollagen, Laminin, and Heparan Sulfate Proteoglycan from the EHS Sarcoma. Biochemistry 1982, 21, 6188–6193. [Google Scholar] [CrossRef]
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef]
- Donaldson, S.S.; Laningham, F.; Fisher, P.G. Advances toward an Understanding of Brainstem Gliomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 1266–1272. [Google Scholar] [CrossRef]
- Johnston, M.; Zakharov, A.; Papaiconomou, C.; Salmasi, G.; Armstrong, D. Evidence of Connections between Cerebrospinal Fluid and Nasal Lymphatic Vessels in Humans, Non-Human Primates and Other Mammalian Species. Cereb. Fluid Res. 2004, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Orešković, D.; Klarica, M. Development of Hydrocephalus and Classical Hypothesis of Cerebrospinal Fluid Hydrodynamics: Facts and Illusions. Prog. Neurobiol. 2011, 94, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Magdoom, K.N.; Brown, A.; Rey, J.; Mareci, T.H.; King, M.A.; Sarntinoranont, M. MRI of Whole Rat Brain Perivascular Network Reveals Role for Ventricles in Brain Waste Clearance. Sci. Rep. 2019, 9, 11480. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Host | Age (Weeks) | Location | Total Cells Inoculated | Volume | Suspension Matrix | Days Before Treatment | Treatment Strategy | Treatment Efficiency | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Athymic nude | n.d. | Brainstem | 5 × 105 | n.d. | n.d. | 21 | RG7388 | Enhanced survival | [5] |
| NOD-SCID | 7 | Pons | 3 × 105 | 2 µL | Matrigel | 28 | Panobinostat | No | [25] |
| NOD-SCID | 5 | Pons | 2 × 105 | 5 µL | Matrigel | None | None | Not assessed | [31] |
| Athymic nude | 6 | Pons | 5 × 105 | 5 µL | PBS | 37 | Doxorubicin and FUS | No | [32] |
| Athymic nude | n.d. | Pons | 5 × 105 | 5 µL | n.d. | 14 | BGB324, Panobinostat and CED | Enhanced survival combined with CED | [37] |
| NOD-SCID | 6–8 | 4th Ventricle | 5 × 105 | 4 µL | Matrigel | 28 | OKN-007 and LDN-193189 | Reduced cellular activity | [38] |
| NOD-SCID gamma (NSG) | 8–10 | Pons | 4 × 105 | 2 µL | Medium:Matrigel (1:1) | 21 | 2-DG and IDH1 inhibitor | Enhanced survival and decreased growth | [39] |
| Athymic nude rat | 4 | 4th Ventricle | 7.5 × 105 | 7.5 µL | n.d. | 28 | SN-38 | Not assessed | [40] |
| Nude BALB/c | 8 | 4th Ventricle/Pons | 2 × 105 | 3 µL | n.d. | 21 | DCA, Metformin and RT | Enhanced survival combined with RT | [41] |
| NOD-SCID | 7–8 | 4th Ventricle/Pons | 2 × 105 | 2 µL | Matrigel | 28 | CBL0137 and Panobinostat | Enhanced survival in combination | [42] |
| Nude BALB/c | 6 | Pons | 1 × 104 | 1 µL | HBSS | 21 | CRAd.S.pK7 | Enhanced survival | [43] |
| NOD-SCID gamma (NSG) | 5–7 | Pons | 1 × 105 | 2 µL | Serum free media | 80 | ALDH+/− and GDC-0084 | Enhanced survival | [44] |
| Nude BALB/c | 5–7 | Brainstem | 2 × 105 | 2 µL | Matrigel | 30 | DMFO & AMXT | Enhanced survival and decreased growth | [45] |
| Athymic nude | 3 | 4th Ventricle | 5 × 105 | 5 µL | Matrigel | 25 | Vandetanib and Everolimus | Enhanced survival in combination | [46] |
| Athymic nude | 7–9 | Striatum/Pons | 5 × 105 | 5 µL | PBS | 75 | Bevacizumab | Not assessed | [47] |
| Athymic nude | 6–8 | Pons | 5 × 105 | 5 µL | PBS | 7–8 | Doxorubicin and CED | No | [48] |
| NOD-SCID | 4–5 | 4th Ventricle | 5 × 105 | 2 µL | PBS | 0 | HSV1716 | Reduced cellular growth | [49] |
| Nude BALB/c | 5–6 | 4th Ventricle/Pons | 2 × 105 | 2 µL | Matrigel | 28–35 | Temozolomide and RT | RT enhanced survival | [50] |
| NOD-SCID gamma (NSG) | 0–2 days | Brainstem | 1 × 103 | n.d. | Tumour stem medium | 0 | GSK2830371 | Enhanced survival | [51] |
| NOD-SCID | 3 | Pons | 5 × 105 | 5 µL | Matrigel | 21–28 | LDN-193189 and LDN-214117 | Enhanced survival | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
’t Hart, E.; Bianco, J.; Besse, H.C.; Chin Joe Kie, L.A.; Cornet, L.; Eikelenboom, K.L.; van den Broek, T.J.M.; Derieppe, M.; Su, Y.; Hoving, E.W.; et al. Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research. Biomedicines 2023, 11, 527. https://doi.org/10.3390/biomedicines11020527
’t Hart E, Bianco J, Besse HC, Chin Joe Kie LA, Cornet L, Eikelenboom KL, van den Broek TJM, Derieppe M, Su Y, Hoving EW, et al. Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research. Biomedicines. 2023; 11(2):527. https://doi.org/10.3390/biomedicines11020527
Chicago/Turabian Style’t Hart, Elvin, John Bianco, Helena C. Besse, Lois A. Chin Joe Kie, Lesley Cornet, Kimberly L. Eikelenboom, Thijs J.M. van den Broek, Marc Derieppe, Yan Su, Eelco W. Hoving, and et al. 2023. "Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research" Biomedicines 11, no. 2: 527. https://doi.org/10.3390/biomedicines11020527
APA Style’t Hart, E., Bianco, J., Besse, H. C., Chin Joe Kie, L. A., Cornet, L., Eikelenboom, K. L., van den Broek, T. J. M., Derieppe, M., Su, Y., Hoving, E. W., Ries, M. G., & van Vuurden, D. G. (2023). Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research. Biomedicines, 11(2), 527. https://doi.org/10.3390/biomedicines11020527