

Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies



Abstract
1. Introduction
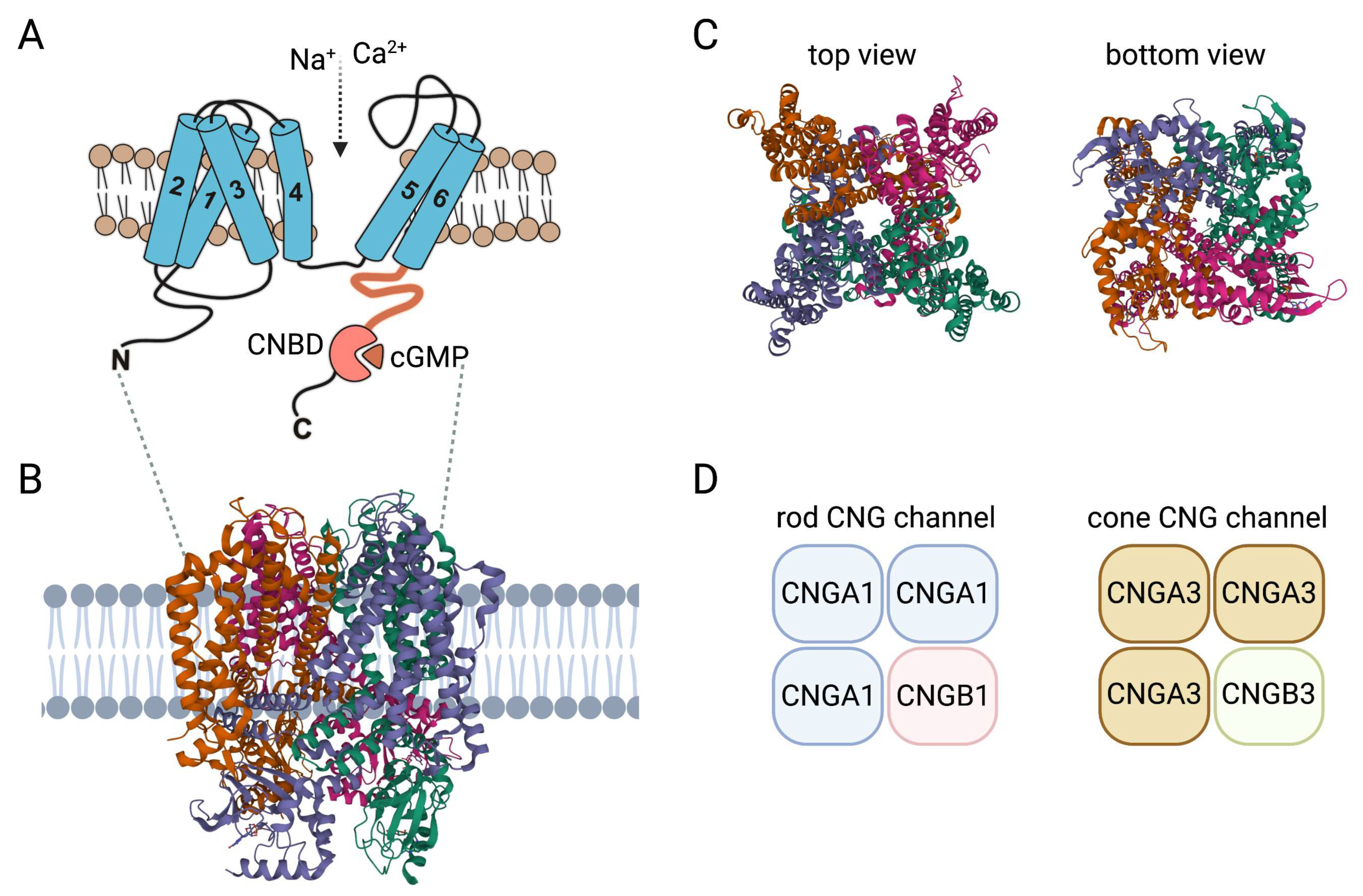
2. Insights on Structure and Activation of CNG Channels
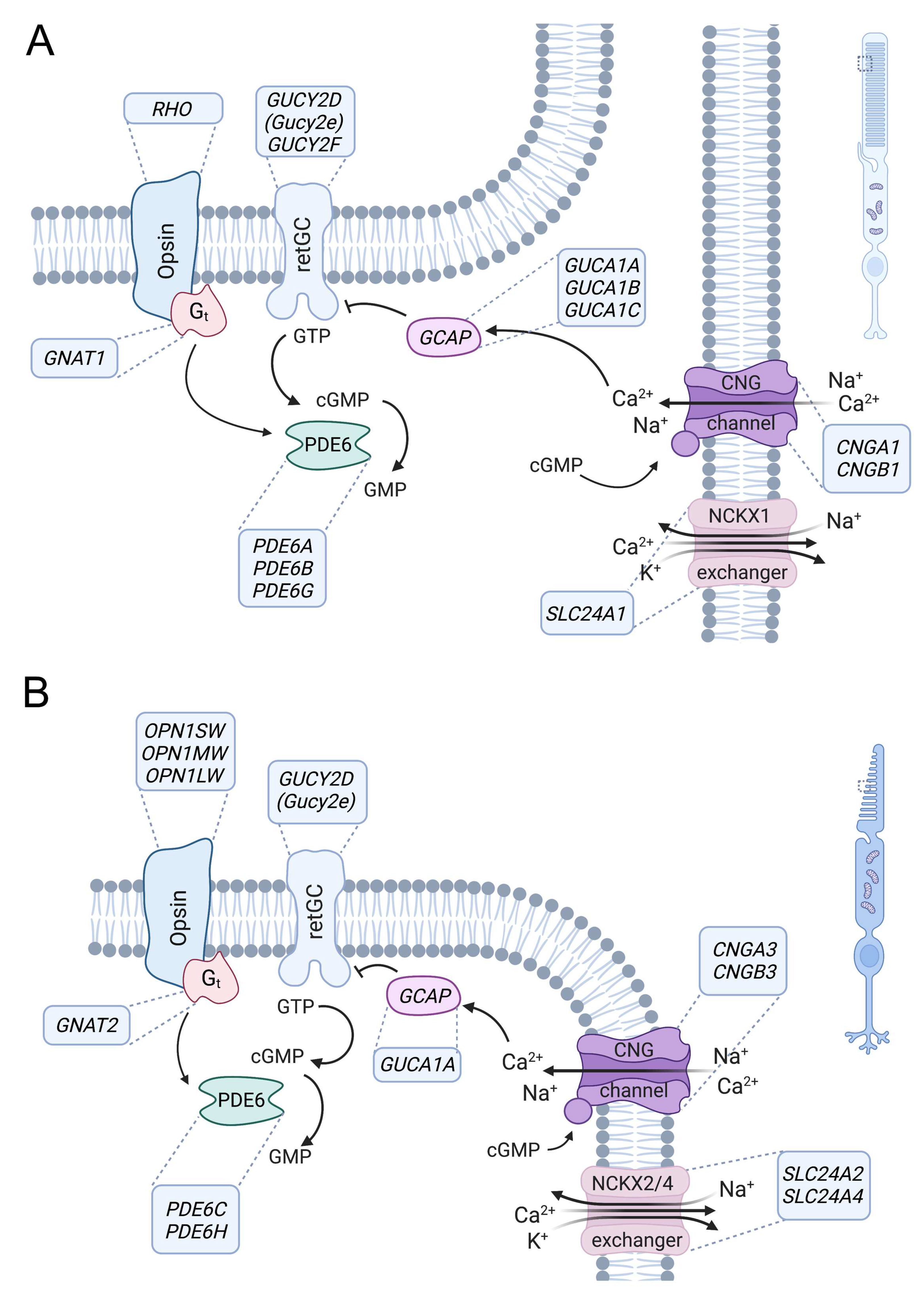
3. Role of CNG Channels in Signal Transduction in Photoreceptors
4. Genetics and Biology of the Rod and Cone CNG Channel
4.1. The Rod CNGA1/CNGB1 Channel
4.2. The Cone CNGA3/CNGB3 Channel

{kind=link}
{kind=link}
| Gene | Chromosomal Location | Phenotype, OMIM | Animal Models | POC Studies | Preclinical Safety Studies | Clinical Trials (NCT ID) |
|---|---|---|---|---|---|---|
| CNGA1 | 4q12 | RP49, 613756 | knockout mouse [53] mutant mouse [63] canine model [52] | - | - | - |
| CNGB1 | 16q21 | RP45, 613767 | knockout mouse [54,56] canine model [55,57,59] | Refs. [59,129,130] | - | - |
| CNGA3 | 2q11.2 | ACHM2, 600053 | knockout mouse [117] mutant mouse [131] canine model [122] ovine model [121] | Refs. [119,132,133,134,135,136,137,138] | Refs. [137,138,139,140,141,142] | 02610582 02935517 03758404 03278873 |
| CNGB3 | 8q21.3 | ACHM3, 605080 | knockout mouse [123] mutant mouse [125] canine model [126] in situ NHP model [128] | Refs. [143,144,145] | Refs. [146,147,148] | 02599922 03001310 03278873 |
5. Gene Therapy for the Treatment of CNG Channelopathies
5.1. The AAV Vector Technology
5.2. Gene Therapy for CNG-Channel-Linked RP
5.3. Gene Therapy for CNG-Channel-Linked ACHM
5.3.1. ACHM Gene Therapy: Preclinical Proof-of-Concept Studies
5.3.2. ACHM Gene Therapy: Clinical Studies
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 191, 111–136. [Google Scholar] [CrossRef]
- Li, M.; Zhou, X.; Wang, S.; Michailidis, I.; Gong, Y.; Su, D.; Li, H.; Li, X.; Yang, J. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 2017, 542, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Han, Y.; Zeng, W.; Jiang, Y. Structural mechanisms of assembly, permeation, gating, and pharmacology of native human rod CNG channel. Neuron 2022, 110, 86–95.e85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hu, Z.; Li, H.; Yang, J. Structure of the human cone photoreceptor cyclic nucleotide-gated channel. Nat. Struct Mol. Biol. 2022, 29, 40–46. [Google Scholar] [CrossRef]
- Zhong, H.; Molday, L.L.; Molday, R.S.; Yau, K.W. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature 2002, 420, 193–198. [Google Scholar] [CrossRef]
- Zheng, J.; Trudeau, M.C.; Zagotta, W.N. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron 2002, 36, 891–896. [Google Scholar] [CrossRef]
- Weitz, D.; Ficek, N.; Kremmer, E.; Bauer, P.J.; Kaupp, U.B. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron 2002, 36, 881–889. [Google Scholar] [CrossRef]
- Shuart, N.G.; Haitin, Y.; Camp, S.S.; Black, K.D.; Zagotta, W.N. Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide-gated ion channels. Nat. Commun. 2011, 2, 457. [Google Scholar] [CrossRef]
- James, Z.M.; Zagotta, W.N. Structural insights into the mechanisms of CNBD channel function. J. Gen. Physiol. 2018, 150, 225–244. [Google Scholar] [CrossRef]
- Zheng, X.; Li, H.; Hu, Z.; Su, D.; Yang, J. Structural and functional characterization of an achromatopsia-associated mutation in a phototransduction channel. Commun. Biol. 2022, 5, 190. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Han, Y.; Zeng, W.; Wang, Y.; Jiang, Y. Structural mechanisms of gating and selectivity of human rod CNGA1 channel. Neuron 2021, 109, 1302–1313.e1304. [Google Scholar] [CrossRef] [PubMed]
- Barret, D.C.A.; Schertler, G.F.X.; Kaupp, U.B.; Marino, J. Structural basis of the partially open central gate in the human CNGA1/CNGB1 channel explained by additional density for calmodulin in cryo-EM map. J. Struct. Biol. 2022, 214, 107828. [Google Scholar] [CrossRef] [PubMed]
- Barret, D.C.A.; Schertler, G.F.X.; Benjamin Kaupp, U.; Marino, J. The structure of the native CNGA1/CNGB1 CNG channel from bovine retinal rods. Nat. Struct. Mol. Biol. 2022, 29, 32–39. [Google Scholar] [CrossRef]
- Zhou, L.; Siegelbaum, S.A. Gating of HCN Channels by cyclic nucleotides: Residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 2007, 15, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, W.N.; Olivier, N.B.; Black, K.D.; Young, E.C.; Olson, R.; Gouaux, E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 2003, 425, 200–205. [Google Scholar] [CrossRef]
- Taraska, J.W.; Zagotta, W.N. Cyclic nucleotide-regulated ion channels: Spotlight on symmetry. Structure 2007, 15, 1023–1024. [Google Scholar] [CrossRef]
- Flynn, G.E.; Black, K.D.; Islas, L.D.; Sankaran, B.; Zagotta, W.N. Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure 2007, 15, 671–682. [Google Scholar] [CrossRef]
- Napolitano, L.M.R.; Torre, V.; Marchesi, A. CNG channel structure, function, and gating: A tale of conformational flexibility. Pflugers Arch. 2021, 473, 1423–1435. [Google Scholar] [CrossRef]
- Ahnelt, P.K.; Kolb, H. The mammalian photoreceptor mosaic-adaptive design. Prog. Retinal Eye Res. 2000, 19, 711–777. [Google Scholar] [CrossRef]
- Yokoyama, S. Molecular evolution of vertebrate visual pigments. Prog. Retinal Eye Res. 2000, 19, 385–419. [Google Scholar] [CrossRef]
- Pugh, E.N., Jr.; Duda, T.; Sitaramayya, A.; Sharma, R.K. Photoreceptor guanylate cyclases: A review. Biosci Rep. 1997, 17, 429–473. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.B.; Robinson, S.W.; Xiong, W.H.; Yau, K.W.; Birch, D.G.; Garbers, D.L. Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. Journal Neurosci. 1999, 19, 5889–5897. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Getter, T.; Salom, D.; Wu, D.; Quetschlich, D.; Chorev, D.S.; Palczewski, K.; Robinson, C.V. Capturing a rhodopsin receptor signalling cascade across a native membrane. Nature 2022, 604, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; McNaughton, P.A.; Nunn, B.J. The ionic selectivity and calcium dependence of the light-sensitive pathway in toad rods. J. Physiol. 1985, 358, 447–468. [Google Scholar] [CrossRef]
- Yau, K.W.; Nakatani, K. Cation selectivity of light-sensitive conductance in retinal rods. Nature 1984, 309, 352–354. [Google Scholar] [CrossRef]
- Cervetto, L.; Lagnado, L.; Perry, R.J.; Robinson, D.W.; McNaughton, P.A. Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients. Nature 1989, 337, 740–743. [Google Scholar] [CrossRef]
- Schnetkamp, P.P. The SLC24 Na+/Ca2+-K+ exchanger family: Vision and beyond. Pflugers Arch. 2004, 447, 683–688. [Google Scholar] [CrossRef]
- Vinberg, F.; Wang, T.; De Maria, A.; Zhao, H.; Bassnett, S.; Chen, J.; Kefalov, V.J. The Na(+)/Ca(2+), K(+) exchanger NCKX4 is required for efficient cone-mediated vision. eLife 2017, 6, e24550. [Google Scholar] [CrossRef]
- Yau, K.W.; Nakatani, K. Light-induced reduction of cytoplasmic free calcium in retinal rod outer segment. Nature 1985, 313, 579–582. [Google Scholar] [CrossRef]
- Irwin, M.J.; Gupta, R.; Gao, X.Z.; Cahill, K.B.; Chu, F.; Cote, R.H. The molecular architecture of photoreceptor phosphodiesterase 6 (PDE6) with activated G protein elucidates the mechanism of visual excitation. J. Biological Chem. 2019, 294, 19486–19497. [Google Scholar] [CrossRef]
- Gao, Y.; Eskici, G.; Ramachandran, S.; Poitevin, F.; Seven, A.B.; Panova, O.; Skiniotis, G.; Cerione, R.A. Structure of the Visual Signaling Complex between Transducin and Phosphodiesterase 6. Mol. Cell 2020, 80, 237–245.e234. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.B. Structural Insights into Retinal Guanylate Cyclase Activator Proteins (GCAPs). Int. J. Mol. Sci. 2021, 22, 8731. [Google Scholar] [CrossRef]
- Ohyama, T.; Hackos, D.H.; Frings, S.; Hagen, V.; Kaupp, U.B.; Korenbrot, J.I. Fraction of the dark current carried by Ca(2+) through cGMP-gated ion channels of intact rod and cone photoreceptors. J. Gen. Physiol. 2000, 116, 735–754. [Google Scholar] [CrossRef] [PubMed]
- Korenbrot, J.I. Speed, sensitivity, and stability of the light response in rod and cone photoreceptors: Facts and models. Prog. Retin. Eye Res. 2012, 31, 442–466. [Google Scholar] [CrossRef] [PubMed]
- Bareil, C.; Hamel, C.P.; Delague, V.; Arnaud, B.; Demaille, J.; Claustres, M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum. Genet. 2001, 108, 328–334. [Google Scholar] [CrossRef]
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Kalloniatis, M.; Fletcher, E.L. Retinitis pigmentosa: Understanding the clinical presentation, mechanisms and treatment options. Clin. Exp. Optom. 2004, 87, 65–80. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, K.; Sheng, X.; Li, Y.; Gao, X.; Zhang, X.; Kang, X.; Pan, X.; Liu, Y.; Jiang, C.; et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2186–2197. [Google Scholar] [CrossRef]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Akahori, M.; Sergeev, Y.; Yoshitake, K.; Ikeo, K.; Furuno, M.; Hayashi, T.; Kondo, M.; Ueno, S.; Tsunoda, K.; et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PloS ONE 2014, 9, e108721. [Google Scholar] [CrossRef]
- Paloma, E.; Martinez-Mir, A.; Garcia-Sandoval, B.; Ayuso, C.; Vilageliu, L.; Gonzalez-Duarte, R.; Balcells, S. Novel homozygous mutation in the alpha subunit of the rod cGMP gated channel (CNGA1) in two Spanish sibs affected with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2002, 39, E66. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, N.; Ildefonse, M.; Pages, F.; Ragno, M.; Bennett, N. Basis for intracellular retention of a human mutant of the retinal rod channel alpha subunit. J. Membr. Biol. 2002, 185, 129–136. [Google Scholar] [CrossRef]
- Ge, Z.; Bowles, K.; Goetz, K.; Scholl, H.P.; Wang, F.; Wang, X.; Xu, S.; Wang, K.; Wang, H.; Chen, R. NGS-based Molecular diagnosis of 105 eyeGENE((R)) probands with Retinitis Pigmentosa. Sci. Rep. 2015, 5, 18287. [Google Scholar] [CrossRef]
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Said, S.; Condroyer, C.; Antonio, A.; Kuhlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-related rod-cone dystrophy: A mutation review and update. Hum. Mutat 2021, 42, 641–666. [Google Scholar] [CrossRef]
- Kondo, H.; Qin, M.; Mizota, A.; Kondo, M.; Hayashi, H.; Hayashi, K.; Oshima, K.; Tahira, T.; Hayashi, K. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4433–4439. [Google Scholar] [CrossRef]
- Simpson, D.A.; Clark, G.R.; Alexander, S.; Silvestri, G.; Willoughby, C.E. Molecular diagnosis for heterogeneous genetic diseases with targeted high-throughput DNA sequencing applied to retinitis pigmentosa. J. Med. Genet. 2011, 48, 145–151. [Google Scholar] [CrossRef]
- Michalakis, S.; Zong, X.; Becirovic, E.; Hammelmann, V.; Wein, T.; Wanner, K.T.; Biel, M. The glutamic acid-rich protein is a gating inhibitor of cyclic nucleotide-gated channels. J. Neurosci. 2011, 31, 133–141. [Google Scholar] [CrossRef]
- Becirovic, E.; Nakova, K.; Hammelmann, V.; Hennel, R.; Biel, M.; Michalakis, S. The retinitis pigmentosa mutation c.3444+1G>A in CNGB1 results in skipping of exon 32. PloS ONE 2010, 5, e8969. [Google Scholar] [CrossRef] [PubMed]
- Wiik, A.C.; Ropstad, E.O.; Ekesten, B.; Karlstam, L.; Wade, C.M.; Lingaas, F. Progressive retinal atrophy in Shetland sheepdog is associated with a mutation in the CNGA1 gene. Anim Genet. 2015, 46, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Xiao, Y.; Li, X.; Ruan, S.; Luo, X.; Wan, X.; Wang, F.; Sun, X. Retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGA1. FASEB J. 2021, 35, e21859. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.-G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired Channel Targeting and Retinal Degeneration in Mice Lacking the Cyclic Nucleotide-Gated Channel Subunit CNGB1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef]
- Winkler, P.A.; Ekenstedt, K.J.; Occelli, L.M.; Frattaroli, A.V.; Bartoe, J.T.; Venta, P.J.; Petersen-Jones, S.M. A large animal model for CNGB1 autosomal recessive retinitis pigmentosa. PloS ONE 2013, 8, e72229. [Google Scholar] [CrossRef]
- Zhang, Y.; Molday, L.L.; Molday, R.S.; Sarfare, S.S.; Woodruff, M.L.; Fain, G.L.; Kraft, T.W.; Pittler, S.J. Knockout of GARPs and the beta-subunit of the rod cGMP-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci. 2009, 122, 1192–1200. [Google Scholar] [CrossRef]
- Ahonen, S.J.; Arumilli, M.; Lohi, H. A CNGB1 frameshift mutation in Papillon and Phalene dogs with progressive retinal atrophy. PloS ONE 2013, 8, e72122. [Google Scholar] [CrossRef]
- Winkler, P.A.; Occelli, L.M.; Petersen-Jones, S.M. Large Animal Models of Inherited Retinal Degenerations: A Review. Cells 2020, 9, 882. [Google Scholar] [CrossRef]
- Petersen-Jones, S.M.; Occelli, L.M.; Winkler, P.A.; Lee, W.; Sparrow, J.R.; Tsukikawa, M.; Boye, S.L.; Chiodo, V.; Capasso, J.E.; Becirovic, E.; et al. Patients and animal models of CNGbeta1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Investig. 2018, 128, 190–206. [Google Scholar] [CrossRef]
- Blank, T.; Goldmann, T.; Koch, M.; Amann, L.; Schon, C.; Bonin, M.; Pang, S.; Prinz, M.; Burnet, M.; Wagner, J.E.; et al. Early Microglia Activation Precedes Photoreceptor Degeneration in a Mouse Model of CNGB1-Linked Retinitis Pigmentosa. Front. Immunol. 2017, 8, 1930. [Google Scholar] [CrossRef]
- DeRamus, M.L.; Stacks, D.A.; Zhang, Y.; Huisingh, C.E.; McGwin, G.; Pittler, S.J. GARP2 accelerates retinal degeneration in rod cGMP-gated cation channel beta-subunit knockout mice. Sci. Rep. 2017, 7, 42545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rubin, G.R.; Fineberg, N.; Huisingh, C.; McGwin, G.; Pittler, S.J.; Kraft, T.W. Age-related changes in Cngb1-X1 knockout mice: Prolonged cone survival. Doc. Ophthalmol 2012, 124, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kandaswamy, S.; Zobel, L.; John, B.; Santhiya, S.T.; Bogedein, J.; Przemeck, G.K.H.; Gailus-Durner, V.; Fuchs, H.; Biel, M.; de Angelis, M.H.; et al. Mutations within the cGMP-binding domain of CNGA1 causing autosomal recessive retinitis pigmentosa in human and animal model. Cell Death Discov. 2022, 8, 387. [Google Scholar] [CrossRef]
- Ardell, M.D.; Bedsole, D.L.; Schoborg, R.V.; Pittler, S.J. Genomic organization of the human rod photoreceptor cGMP-gated cation channel beta-subunit gene. Gene 2000, 245, 311–318. [Google Scholar] [CrossRef]
- Batra-Safferling, R.; Abarca-Heidemann, K.; Korschen, H.G.; Tziatzios, C.; Stoldt, M.; Budyak, I.; Willbold, D.; Schwalbe, H.; Klein-Seetharaman, J.; Kaupp, U.B. Glutamic acid-rich proteins of rod photoreceptors are natively unfolded. J. Biological Chem. 2006, 281, 1449–1460. [Google Scholar] [CrossRef]
- Ritter, L.M.; Khattree, N.; Tam, B.; Moritz, O.L.; Schmitz, F.; Goldberg, A.F. In situ visualization of protein interactions in sensory neurons: Glutamic acid-rich proteins (GARPs) play differential roles for photoreceptor outer segment scaffolding. J. Neurosci. 2011, 31, 11231–11243. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; DeRamus, M.L.; Pittler, S.J.; Naash, M.I. Varying the GARP2-to-RDS Ratio Leads to Defects in Rim Formation and Rod and Cone Function. Investig. Ophthalmology Visual Sci. 2015, 56, 8187–8198. [Google Scholar] [CrossRef] [PubMed]
- Pearring, J.N.; Martinez-Marquez, J.; Willer, J.R.; Lieu, E.C.; Salinas, R.Y.; Arshavsky, V.Y. The GARP Domain of the Rod CNG Channel’s beta1-Subunit Contains Distinct Sites for Outer Segment Targeting and Connecting to the Photoreceptor Disk Rim. J. Neurosci. 2021, 41, 3094–3104. [Google Scholar] [CrossRef]
- Ba-Abbad, R.; Holder, G.E.; Robson, A.G.; Neveu, M.M.; Waseem, N.; Arno, G.; Webster, A.R. Isolated rod dysfunction associated with a novel genotype of CNGB1. Am. J. Ophthalmol Case Rep. 2019, 14, 83–86. [Google Scholar] [CrossRef]
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Achromatopsia: Genetics and Gene Therapy. Mol. Diagn Ther 2022, 26, 51–59. [Google Scholar] [CrossRef]
- Neitz, J.; Neitz, M. The genetics of normal and defective color vision. Vision Res. 2011, 51, 633–651. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, R.; Karali, M.; Melillo, P.; Di Iorio, V.; De Benedictis, A.; Iaccarino, G.; Testa, F.; Banfi, S.; Simonelli, F. Clinical and Molecular Characterization of Achromatopsia Patients: A Longitudinal Study. Int J. Mol. Sci 2021, 22, 1681. [Google Scholar] [CrossRef]
- Andreasson, S.; Tornqvist, K. Electroretinograms in patients with achromatopsia. Acta Ophthalmol. 1991, 69, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Felden, J.; Baumann, B.; Ali, M.; Audo, I.; Ayuso, C.; Bocquet, B.; Casteels, I.; Garcia-Sandoval, B.; Jacobson, S.G.; Jurklies, B.; et al. Mutation spectrum and clinical investigation of achromatopsia patients with mutations in the GNAT2 gene. Hum. Mutat 2019, 40, 1145–1155. [Google Scholar] [CrossRef]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jagle, H.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Salati, R.; et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.K.; Van Cauwenbergh, C.; Rother, C.; Baumann, B.; Reuter, P.; De Baere, E.; Wissinger, B.; Kohl, S.; Group, A.S. CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat 2017, 38, 1579–1591. [Google Scholar] [CrossRef]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Inherited retinal diseases: Therapeutics, clinical trials and end points-A review. Clin. Exp. Ophthalmol. 2021, 49, 270–288. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Sun, W.; Li, S.; Xiao, X.; Wang, P.; Zhang, Q. Genotypes and phenotypes of genes associated with achromatopsia: A reference for clinical genetic testing. Molecular vision 2020, 26, 588–602. [Google Scholar]
- Solaki, M.; Baumann, B.; Reuter, P.; Andreasson, S.; Audo, I.; Ayuso, C.; Balousha, G.; Benedicenti, F.; Birch, D.; Bitoun, P.; et al. Comprehensive variant spectrum of the CNGA3 gene in patients affected by achromatopsia. Hum. Mutat 2022, 43, 832–858. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nature genetics 1998, 19, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Slingerland, N.W.; Roosing, S.; van Schooneveld, M.J.; van Lith-Verhoeven, J.J.; van Moll-Ramirez, N.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.; Klaver, C.C. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology 2009, 116, 1984–1989.e1981. [Google Scholar] [CrossRef] [PubMed]
- Wissinger, B.; Gamer, D.; Jagle, H.; Giorda, R.; Marx, T.; Mayer, S.; Tippmann, S.; Broghammer, M.; Jurklies, B.; Rosenberg, T.; et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 2001, 69, 722–737. [Google Scholar] [CrossRef]
- Tränkner, D.; Jagle, H.; Kohl, S.; Apfelstedt-Sylla, E.; Sharpe, L.T.; Kaupp, U.B.; Zrenner, E.; Seifert, R.; Wissinger, B. Molecular basis of an inherited form of incomplete achromatopsia. J. Neurosci. 2004, 24, 138–147. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Sandberg, M.A.; Gorji, N.; Berson, E.L.; Dryja, T.P. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum. Mutat 2005, 25, 248–258. [Google Scholar] [CrossRef]
- Varsanyi, B.; Wissinger, B.; Kohl, S.; Koeppen, K.; Farkas, A. Clinical and genetic features of Hungarian achromatopsia patients. Molecular vision 2005, 11, 996–1001. [Google Scholar]
- Goto-Omoto, S.; Hayashi, T.; Gekka, T.; Kubo, A.; Takeuchi, T.; Kitahara, K. Compound heterozygous CNGA3 mutations (R436W, L633P) in a Japanese patient with congenital achromatopsia. Vis. Neurosci 2006, 23, 395–402. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Lewis, R.A.; Lupski, J.R. Achromatopsia: The CNGB3 p.T383fsX mutation results from a founder effect and is responsible for the visual phenotype in the original report of uniparental disomy 14. Hum. Genet. 2007, 121, 433–439. [Google Scholar] [CrossRef]
- Sundin, O.H.; Yang, J.M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.D.; Maumenee, I.H. Genetic basis of total colourblindness among the Pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jagle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Aligianis, I.A.; Ainsworth, J.R.; Good, P.; Mollon, J.D.; Maher, E.R.; Moore, A.T.; Hunt, D.M. Progressive cone dystrophy associated with mutation in CNGB3. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1975–1982. [Google Scholar]
- Okada, A.; Ueyama, H.; Toyoda, F.; Oda, S.; Ding, W.G.; Tanabe, S.; Yamade, S.; Matsuura, H.; Ohkubo, I.; Kani, K. Functional role of hCngb3 in regulation of human cone cng channel: Effect of rod monochromacy-associated mutations in hCNGB3 on channel function. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2324–2332. [Google Scholar] [CrossRef]
- Rojas, C.V.; Maria, L.S.; Santos, J.L.; Cortes, F.; Alliende, M.A. A frameshift insertion in the cone cyclic nucleotide gated cation channel causes complete achromatopsia in a consanguineous family from a rural isolate. Eur J. Hum. Genet. 2002, 10, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.A.; Hussels, I.; Brink, E.; Torres, J. Hereditary blindness among Pingelapese people of Eastern Caroline Islands. Lancet 1970, 1, 1253–1257. [Google Scholar] [CrossRef]
- Sacks, O.W. The Island of the Colorblind, 1st ed.; A.A. Knopf: New York, NY, USA, 1997; p. 298. [Google Scholar]
- Winick, J.D.; Blundell, M.L.; Galke, B.L.; Salam, A.A.; Leal, S.M.; Karayiorgou, M. Homozygosity mapping of the Achromatopsia locus in the Pingelapese. Am. J. Human Gene. 1999, 64, 1679–1685. [Google Scholar] [CrossRef]
- Zelinger, L.; Cideciyan, A.V.; Kohl, S.; Schwartz, S.B.; Rosenmann, A.; Eli, D.; Sumaroka, A.; Roman, A.J.; Luo, X.; Brown, C.; et al. Genetics and Disease Expression in the CNGA3 Form of Achromatopsia: Steps on the Path to Gene Therapy. Ophthalmology 2015, 122, 997–1007. [Google Scholar] [CrossRef]
- Liang, X.; Dong, F.; Li, H.; Li, H.; Yang, L.; Sui, R. Novel CNGA3 mutations in Chinese patients with achromatopsia. Br. J. Ophthalmol. 2015, 99, 571–576. [Google Scholar] [CrossRef]
- Burkard, M.; Kohl, S.; Kratzig, T.; Tanimoto, N.; Brennenstuhl, C.; Bausch, A.E.; Junger, K.; Reuter, P.; Sothilingam, V.; Beck, S.C.; et al. Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel-associated retinopathy. J. Clinical Investig. 2018, 128, 5663–5675. [Google Scholar] [CrossRef]
- Täger, J.; Wissinger, B.; Kohl, S.; Reuter, P. Identification of Chemical and Pharmacological Chaperones for Correction of Trafficking-Deficient Mutant Cyclic Nucleotide-Gated A3 Channels. Mol. Pharmacol 2021, 99, 460–468. [Google Scholar] [CrossRef]
- Brennenstuhl, C.; Tanimoto, N.; Burkard, M.; Wagner, R.; Bolz, S.; Trifunovic, D.; Kabagema-Bilan, C.; Paquet-Durand, F.; Beck, S.C.; Huber, G.; et al. Targeted ablation of the Pde6h gene in mice reveals cross-species differences in cone and rod phototransduction protein isoform inventory. J. Biol. Chem. 2015, 290, 10242–10255. [Google Scholar] [CrossRef] [PubMed]
- Buena-Atienza, E.; Ruther, K.; Baumann, B.; Bergholz, R.; Birch, D.; De Baere, E.; Dollfus, H.; Greally, M.T.; Gustavsson, P.; Hamel, C.P.; et al. De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy. Sci. Rep. 2016, 6, 28253. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Varnum, M.D. CNGA3 achromatopsia-associated mutation potentiates the phosphoinositide sensitivity of cone photoreceptor CNG channels by altering intersubunit interactions. Am. J. Physiol. Cell Physiol. 2013, 305, C147–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duricka, D.L.; Brown, R.L.; Varnum, M.D. Defective trafficking of cone photoreceptor CNG channels induces the unfolded protein response and ER-stress-associated cell death. Biochem J. 2012, 441, 685–696. [Google Scholar] [CrossRef]
- Koeppen, K.; Reuter, P.; Kohl, S.; Baumann, B.; Ladewig, T.; Wissinger, B. Functional analysis of human CNGA3 mutations associated with colour blindness suggests impaired surface expression of channel mutants A3(R427C) and A3(R563C). Eur. J. Neurosci. 2008, 27, 2391–2401. [Google Scholar] [CrossRef]
- Koeppen, K.; Reuter, P.; Ladewig, T.; Kohl, S.; Baumann, B.; Jacobson, S.G.; Plomp, A.S.; Hamel, C.P.; Janecke, A.R.; Wissinger, B. Dissecting the pathogenic mechanisms of mutations in the pore region of the human cone photoreceptor cyclic nucleotide-gated channel. Hum. Mutat. 2010, 31, 830–839. [Google Scholar] [CrossRef]
- Kuniyoshi, K.; Muraki-Oda, S.; Ueyama, H.; Toyoda, F.; Sakuramoto, H.; Ogita, H.; Irifune, M.; Yamamoto, S.; Nakao, A.; Tsunoda, K.; et al. Novel mutations in the gene for alpha-subunit of retinal cone cyclic nucleotide-gated channels in a Japanese patient with congenital achromatopsia. Jpn J. Ophthalmol. 2016, 60, 187–197. [Google Scholar] [CrossRef]
- Liu, C.; Varnum, M.D. Functional consequences of progressive cone dystrophy-associated mutations in the human cone photoreceptor cyclic nucleotide-gated channel CNGA3 subunit. Am. J. Physiol. Cell Physiol. 2005, 289, C187–C198. [Google Scholar] [CrossRef]
- Matveev, A.V.; Fitzgerald, J.B.; Xu, J.; Malykhina, A.P.; Rodgers, K.K.; Ding, X.Q. The disease-causing mutations in the carboxyl terminus of the cone cyclic nucleotide-gated channel CNGA3 subunit alter the local secondary structure and interfere with the channel active conformational change. Biochemistry 2010, 49, 1628–1639. [Google Scholar] [CrossRef]
- Meighan, P.C.; Peng, C.; Varnum, M.D. Inherited macular degeneration-associated mutations in CNGB3 increase the ligand sensitivity and spontaneous open probability of cone cyclic nucleotide-gated channels. Front. Physiol. 2015, 6, 177. [Google Scholar] [CrossRef]
- Muraki-Oda, S.; Toyoda, F.; Okada, A.; Tanabe, S.; Yamade, S.; Ueyama, H.; Matsuura, H.; Ohji, M. Functional analysis of rod monochromacy-associated missense mutations in the CNGA3 subunit of the cone photoreceptor cGMP-gated channel. Biochem. Biophys. Res. Commun. 2007, 362, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.A.; Bartoli, K.M.; Fandino, R.A.; Ngatchou, A.N.; Woch, G.; Carey, J.; Tanaka, J.C. Transmembrane S1 mutations in CNGA3 from achromatopsia 2 patients cause loss of function and impaired cellular trafficking of the cone CNG channel. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Reuter, P.; Koeppen, K.; Ladewig, T.; Kohl, S.; Baumann, B.; Wissinger, B.; Achromatopsia Clinical Study, G. Mutations in CNGA3 impair trafficking or function of cone cyclic nucleotide-gated channels, resulting in achromatopsia. Hum. Mutat. 2008, 29, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.S.; Reuter, P.; Sisk, R.A.; Kausar, T.; Shahzad, M.; Maqsood, M.I.; Yousif, A.; Ali, M.; Riazuddin, S.; Wissinger, B.; et al. Homozygous missense variant in the human CNGA3 channel causes cone-rod dystrophy. Eur J. Hum. Genet. 2015, 23, 473–480. [Google Scholar] [CrossRef]
- Täger, J.; Kohl, S.; Birch, D.G.; Wheaton, D.K.H.; Wissinger, B.; Reuter, P. An early nonsense mutation facilitates the expression of a short isoform of CNGA3 by alternative translation initiation. Exp. Eye Res. 2018, 171, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Seeliger, M.; Pfeifer, A.; Kohler, K.; Gerstner, A.; Ludwig, A.; Jaissle, G.; Fauser, S.; Zrenner, E.; Hofmann, F. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc. Natl. Acad. Sci. USA 1999, 96, 7553–7557. [Google Scholar] [CrossRef]
- Michalakis, S.; Geiger, H.; Haverkamp, S.; Hofmann, F.; Gerstner, A.; Biel, M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1516–1524. [Google Scholar] [CrossRef]
- Pang, J.J.; Deng, W.T.; Dai, X.; Lei, B.; Everhart, D.; Umino, Y.; Li, J.; Zhang, K.; Mao, S.; Boye, S.L.; et al. AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS ONE 2012, 7, e35250. [Google Scholar] [CrossRef]
- Reicher, S.; Seroussi, E.; Gootwine, E. A mutation in gene CNGA3 is associated with day blindness in sheep. Genomics 2010, 95, 101–104. [Google Scholar] [CrossRef]
- Shamir, M.H.; Ofri, R.; Bor, A.; Brenner, O.; Reicher, S.; Obolensky, A.; Averbukh, E.; Banin, E.; Gootwine, E. A novel day blindness in sheep: Epidemiological, behavioural, electrophysiological and histopathological studies. Vet. J. 2010, 185, 130–137. [Google Scholar] [CrossRef]
- Tanaka, N.; Dutrow, E.V.; Miyadera, K.; Delemotte, L.; MacDermaid, C.M.; Reinstein, S.L.; Crumley, W.R.; Dixon, C.J.; Casal, M.L.; Klein, M.L.; et al. Canine CNGA3 Gene Mutations Provide Novel Insights into Human Achromatopsia-Associated Channelopathies and Treatment. PloS ONE 2015, 10, e0138943. [Google Scholar] [CrossRef]
- Ding, X.Q.; Harry, C.S.; Umino, Y.; Matveev, A.V.; Fliesler, S.J.; Barlow, R.B. Impaired cone function and cone degeneration resulting from CNGB3 deficiency: Down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum. Mol. Genet. 2009, 18, 4770–4780. [Google Scholar] [CrossRef]
- Xu, J.; Morris, L.; Fliesler, S.J.; Sherry, D.M.; Ding, X.Q. Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3557–3566. [Google Scholar] [CrossRef][Green Version]
- Hassall, M.M.; Barnard, A.R.; Orlans, H.O.; McClements, M.E.; Charbel Issa, P.; Aslam, S.A.; MacLaren, R.E. A Novel Achromatopsia Mouse Model Resulting From a Naturally Occurring Missense Change in Cngb3. Investig. Ophthalmol. Vis. Sci. 2018, 59, 6102–6110. [Google Scholar] [CrossRef]
- Sidjanin, D.J.; Lowe, J.K.; McElwee, J.L.; Milne, B.S.; Phippen, T.M.; Sargan, D.R.; Aguirre, G.D.; Acland, G.M.; Ostrander, E.A. Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum. Mol. Genet. 2002, 11, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, G.D.; Rubin, L.F. Pathology of hemeralopia in the Alaskan malamute dog. Investig. Ophthalmol. 1974, 13, 231–235. [Google Scholar] [PubMed]
- Lin, Q.; Lv, J.N.; Wu, K.C.; Zhang, C.J.; Liu, Q.; Jin, Z.B. Generation of Nonhuman Primate Model of Cone Dysfunction through In Situ AAV-Mediated CNGB3 Ablation. Mol. Ther. Methods Clin. Dev. 2020, 18, 869–879. [Google Scholar] [CrossRef]
- Koch, S.; Sothilingam, V.; Garcia Garrido, M.; Tanimoto, N.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; Seeliger, M.W.; Biel, M.; et al. Gene therapy restores vision and delays degeneration in the CNGB1(-/-) mouse model of retinitis pigmentosa. Hum. Mol. Genet. 2012, 21, 4486–4496. [Google Scholar] [CrossRef]
- Wagner, J.E.; Zobel, L.; Gerhardt, M.J.; O’Riordan, C.R.; Frederick, A.; Petersen-Jones, S.M.; Biel, M.; Michalakis, S. In Vivo Potency Testing of Subretinal rAAV5.hCNGB1 Gene Therapy in the Cngb1 Knockout Mouse Model of Retinitis Pigmentosa. Hum. Gene Ther. 2021, 32, 1158–1170. [Google Scholar] [CrossRef]
- Hawes, N.; Wang, X.; Hurd, R.; Wang, J.; Davisson, M.; Nusinowitz, S.; Heckenlively, J.; Chang, B. A point mutation in the Cnga3 gene causes cone photoreceptor function loss (cpfl5) in mice. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4579. [Google Scholar]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Mühlfriedel, R.; Tanimoto, N.; Schön, C.; Sothilingam, V.; Garcia Garrido, M.; Beck, S.C.; Huber, G.; Biel, M.; Seeliger, M.W.; Michalakis, S. AAV-Mediated Gene Supplementation Therapy in Achromatopsia Type 2: Preclinical Data on Therapeutic Time Window and Long-Term Effects. Front. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Tao, Y.; Deng, W.T.; Zhu, P.; Li, J.; Dai, X.; Zhang, Y.; Shi, W.; Liu, X.; Chiodo, V.A.; et al. Vitreal delivery of AAV vectored Cnga3 restores cone function in CNGA3-/-/Nrl-/- mice, an all-cone model of CNGA3 achromatopsia. Hum. Mol. Genet. 2015, 24, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV capsids for intravitreal gene therapy of photoreceptor disorders. EMBO Mol. Med. 2021, 13, e13392. [Google Scholar] [CrossRef]
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Honig, H.; Rosov, A.; Yamin, E.; Sharon, D.; et al. Gene Augmentation Therapy Restores Retinal Function and Visual Behavior in a Sheep Model of CNGA3 Achromatopsia. Mol. Ther. 2015, 23, 1423–1433. [Google Scholar] [CrossRef]
- Gootwine, E.; Ofri, R.; Banin, E.; Obolensky, A.; Averbukh, E.; Ezra-Elia, R.; Ross, M.; Honig, H.; Rosov, A.; Yamin, E.; et al. Safety and Efficacy Evaluation of rAAV2tYF-PR1.7-hCNGA3 Vector Delivered by Subretinal Injection in CNGA3 Mutant Achromatopsia Sheep. Hum. Gene Ther. Clin. Dev. 2017, 28, 96–107. [Google Scholar] [CrossRef]
- Ofri, R.; Averbukh, E.; Ezra-Elia, R.; Ross, M.; Honig, H.; Obolensky, A.; Rosov, A.; Hauswirth, W.W.; Gootwine, E.; Banin, E. Six Years and Counting: Restoration of Photopic Retinal Function and Visual Behavior Following Gene Augmentation Therapy in a Sheep Model of CNGA3 Achromatopsia. Human. Gene Ther. 2018, 29, 1376–1386. [Google Scholar] [CrossRef]
- Reichel, F.F.; Peters, T.; Wilhelm, B.; Biel, M.; Ueffing, M.; Wissinger, B.; Bartz-Schmidt, K.U.; Klein, R.; Michalakis, S.; Fischer, M.D.; et al. Humoral Immune Response After Intravitreal But Not After Subretinal AAV8 in Primates and Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1910–1915. [Google Scholar] [CrossRef]
- Seitz, I.P.; Michalakis, S.; Wilhelm, B.; Reichel, F.F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Biel, M.; Wissinger, B.; Bartz-Schmidt, K.U.; et al. Superior Retinal Gene Transfer and Biodistribution Profile of Subretinal Versus Intravitreal Delivery of AAV8 in Nonhuman Primates. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5792–5801. [Google Scholar] [CrossRef]
- Tobias, P.; Philipp, S.I.; Stylianos, M.; Martin, B.; Barbara, W.; Felix, R.; Alexander, O.G.; Eberhart, Z.; Marius, U.; Birgit, K.; et al. Safety and Toxicology of Ocular Gene Therapy with Recombinant AAV Vector rAAV.hCNGA3 in Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2019, 30, 50–56. [Google Scholar] [CrossRef]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol. Therapy. 2017, 25, 2648–2660. [Google Scholar] [CrossRef]
- Komaromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Komaromy, A.M.; Rowlan, J.S.; Corr, A.T.; Reinstein, S.L.; Boye, S.L.; Cooper, A.E.; Gonzalez, A.; Levy, B.; Wen, R.; Hauswirth, W.W.; et al. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol. Therapy 2013, 21, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Fliesler, S.J.; Ding, X.Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; Sharma, A.K.; Ver Hoeve, J.N.; Smith, L.M.; Arndt, T.; Calcedo, R.; et al. Safety and Biodistribution Evaluation in Cynomolgus Macaques of rAAV2tYF-PR1.7-hCNGB3, a Recombinant AAV Vector for Treatment of Achromatopsia. Hum. Gene Ther Clin. Dev. 2016, 27, 37–48. [Google Scholar] [CrossRef]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; McPherson, L.; Ver Hoeve, J.N.; Smith, L.M.; Arndt, T.; Mandapati, S.; et al. Safety and Biodistribution Evaluation in CNGB3-Deficient Mice of rAAV2tYF-PR1.7-hCNGB3, a Recombinant AAV Vector for Treatment of Achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Komaromy, A.M.; Zeiss, C.; Calcedo, R.; Harman, C.D.; Koehl, K.L.; Stewart, G.A.; Iwabe, S.; Chiodo, V.A.; Hauswirth, W.W.; et al. Safety and Efficacy of AAV5 Vectors Expressing Human or Canine CNGB3 in CNGB3-Mutant Dogs. Hum. Gene Ther. Clin. Dev. 2017, 28, 197–207. [Google Scholar] [CrossRef]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retinal Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef]
- Penaud-Budloo, M.; Francois, A.; Clement, N.; Ayuso, E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther Methods Clin. Dev. 2018, 8, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.; Chan, Y.K.; Samulski, R.J. Adeno-associated Virus (AAV) versus Immune Response. Viruses 2019, 11, 102. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Therapy. 2020, 28, 723–746. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Grimm, D. Adeno-Associated Viruses (AAV) and Host Immunity—A Race Between the Hare and the Hedgehog. Front. Immunol. 2021, 12, 753467. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Seitz, I.P.; Michalakis, S.; Biel, M.; Wilhelm, B.; Reichel, F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Korbmacher, B.; et al. Safety and Toxicology of Ocular Gene Therapy with Recombinant AAV Vector rAAV.hCNGA3 in Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2019, 30, 50–56. [Google Scholar] [CrossRef]
- Reichel, F.F.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Weisschuh, N.; Sothilingam, V.; Kuehlewein, L.; Kahle, N.; et al. Three-year results of phase I retinal gene therapy trial for CNGA3-mutated achromatopsia: Results of a non randomised controlled trial. Bri. J. Ophthalmol. 2021, 106, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.D.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Weisschuh, N.; Ochakovski, G.A.; Klein, R.; Schoen, C.; et al. Safety and Vision Outcomes of Subretinal Gene Therapy Targeting Cone Photoreceptors in Achromatopsia: A Nonrandomized Controlled Trial. JAMA Ophthalmol. 2020, 138, 643–651. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerhardt, M.J.; Priglinger, S.G.; Biel, M.; Michalakis, S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines 2023, 11, 269. https://doi.org/10.3390/biomedicines11020269
Gerhardt MJ, Priglinger SG, Biel M, Michalakis S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines. 2023; 11(2):269. https://doi.org/10.3390/biomedicines11020269
Chicago/Turabian StyleGerhardt, Maximilian J., Siegfried G. Priglinger, Martin Biel, and Stylianos Michalakis. 2023. "Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies" Biomedicines 11, no. 2: 269. https://doi.org/10.3390/biomedicines11020269
APA StyleGerhardt, M. J., Priglinger, S. G., Biel, M., & Michalakis, S. (2023). Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines, 11(2), 269. https://doi.org/10.3390/biomedicines11020269