
VEGFR2 Expression Correlates with Postnatal Development of Brain Arteriovenous Malformations in a Mouse Model of Type I Hereditary Hemorrhagic Telangiectasia

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transgenic Mice and Conditional Gene Deletion
2.2. AVM Visualization Using Latex Dye Perfusion
2.3. Hemoglobin Concentration
2.4. Western Blotting
2.5. Magnetic Resonance Imaging
2.6. X-gal Staining and Vascular Density Quantification
2.7. Statistical Analysis
3. Results
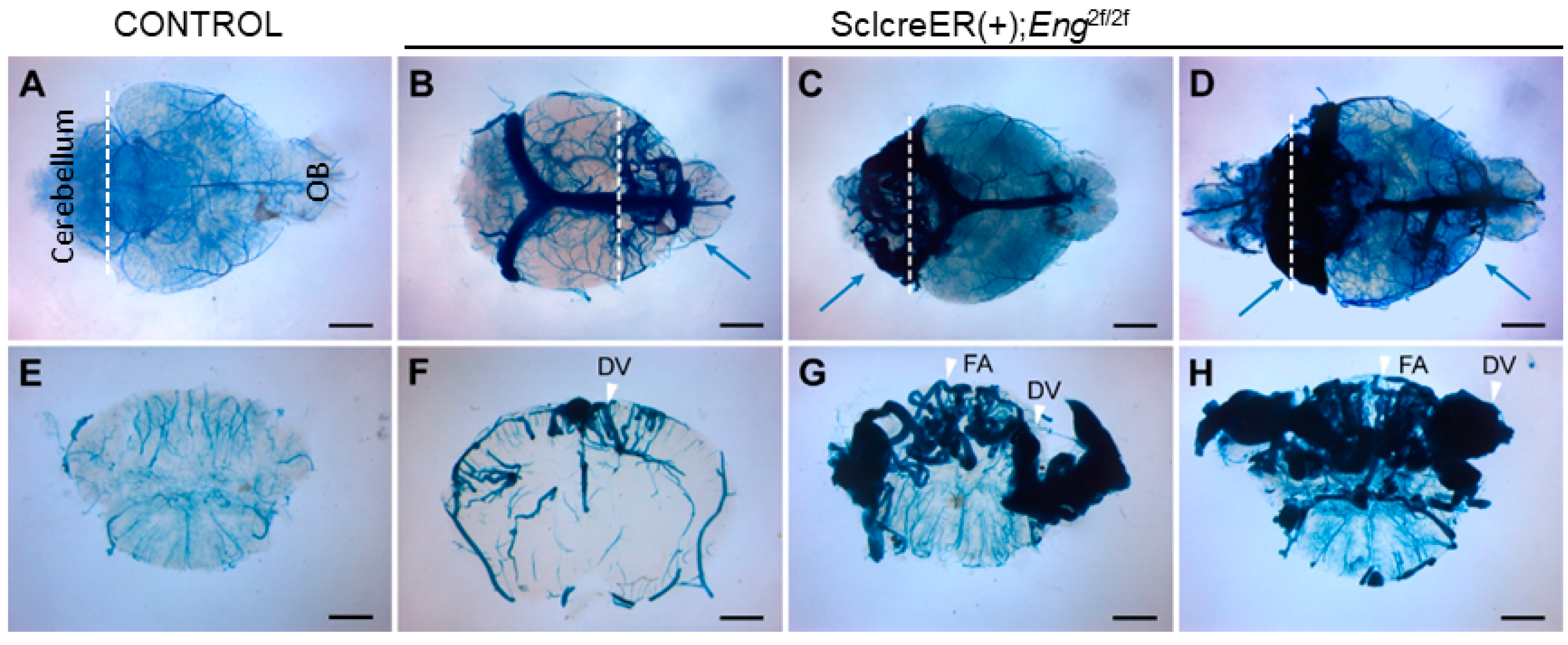
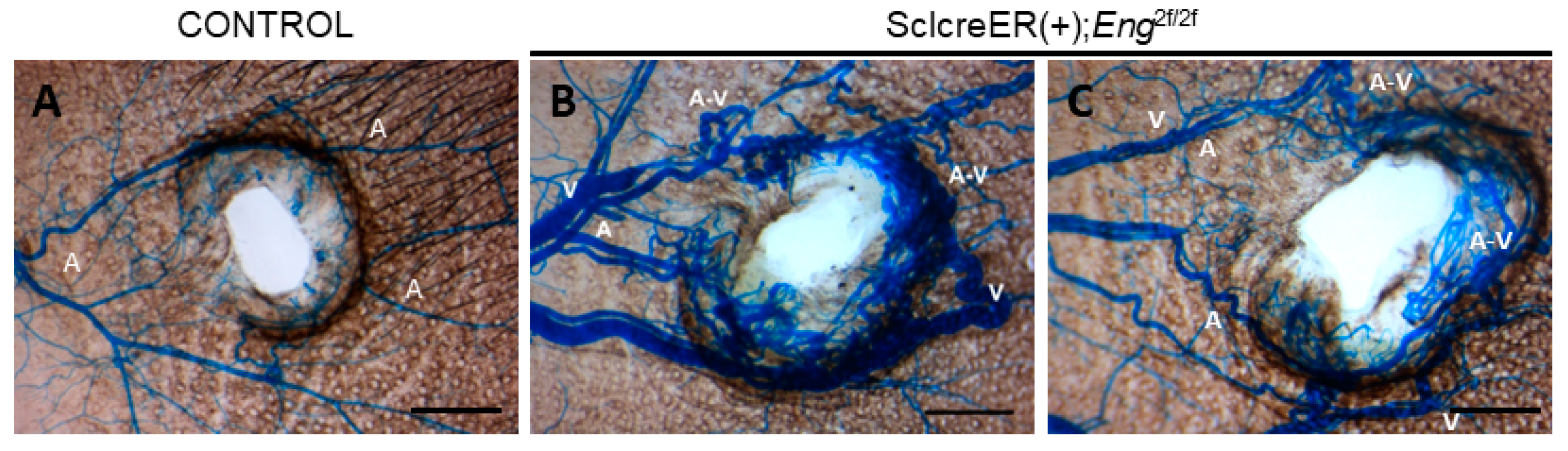
3.1. Endothelial Cell Eng Gene Deletion Induced BAVM Development
3.2. Longitudinal Monitoring of BAVMs Using Magnetic Resonance Angiography
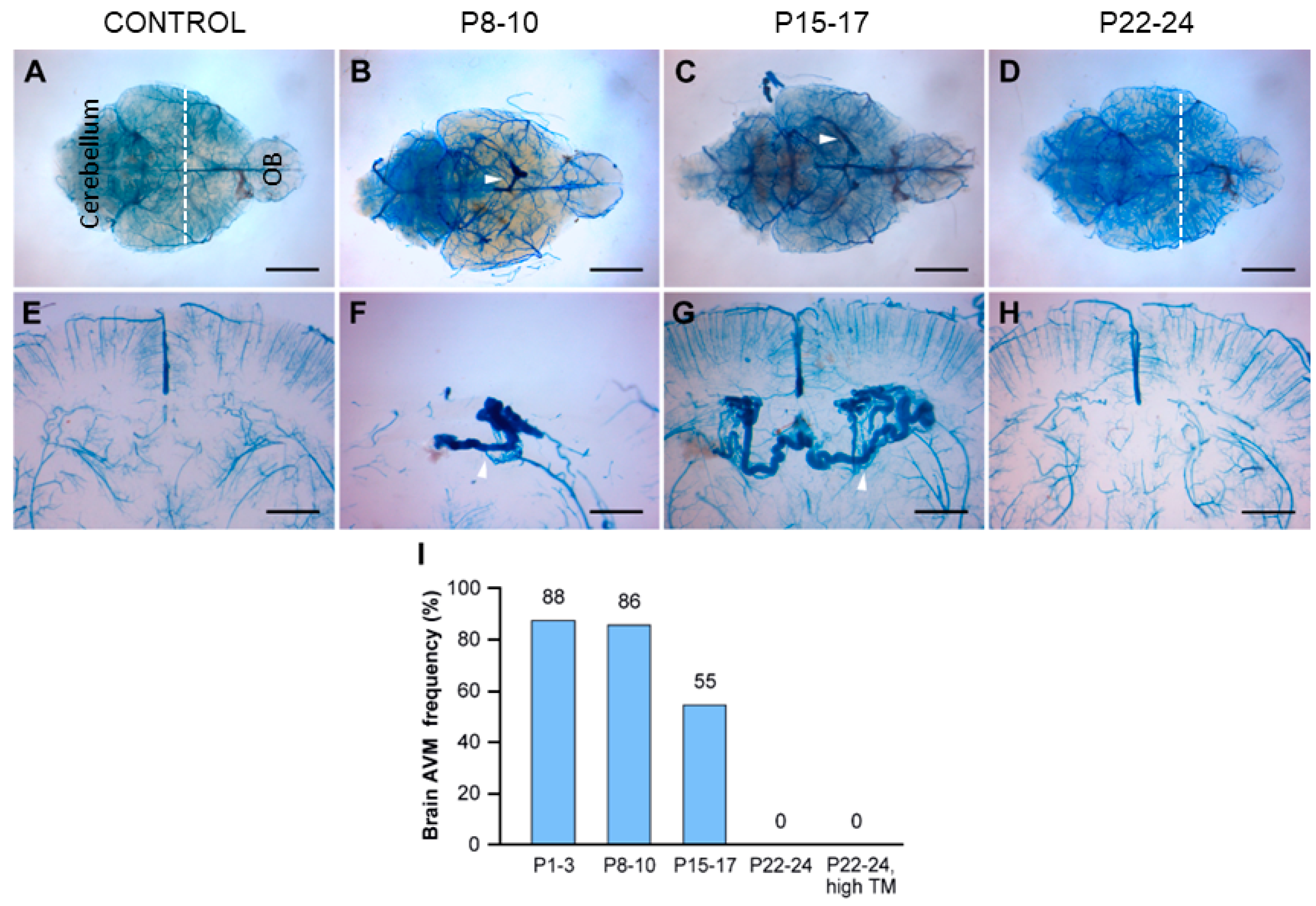
3.3. The Early Postnatal Period Was Critical for BAVM Development
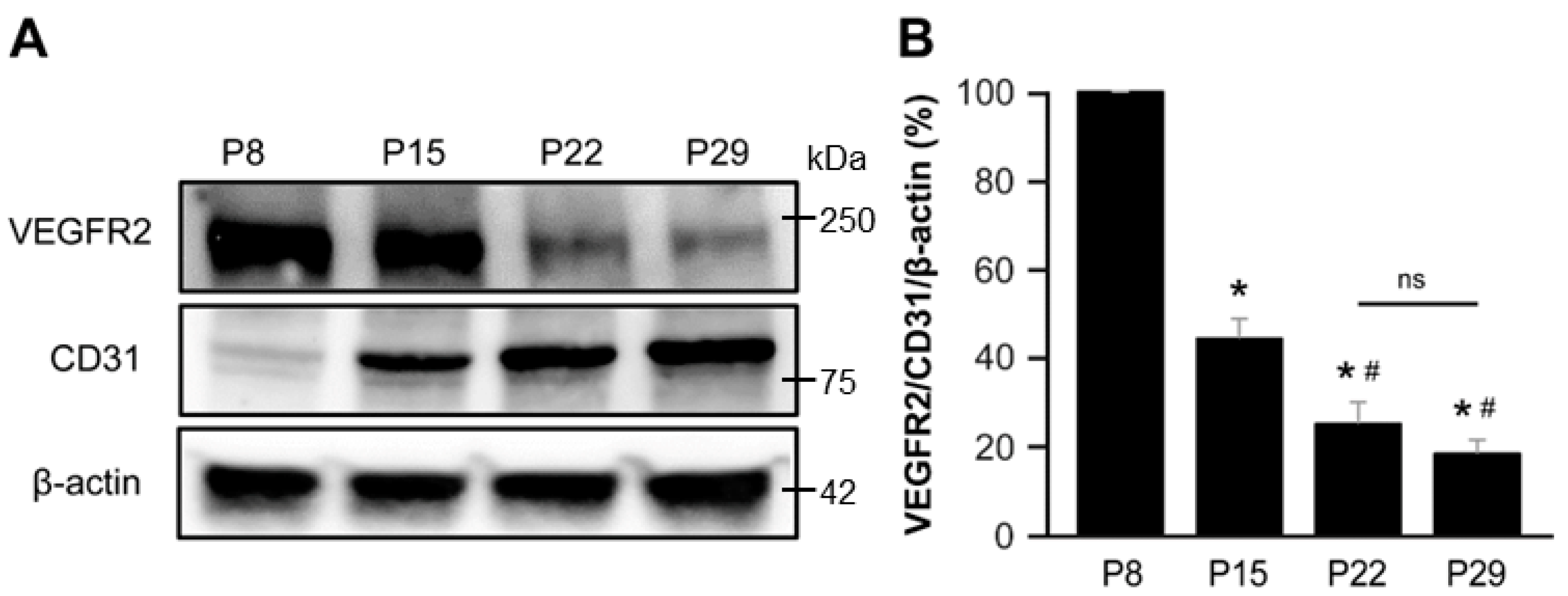
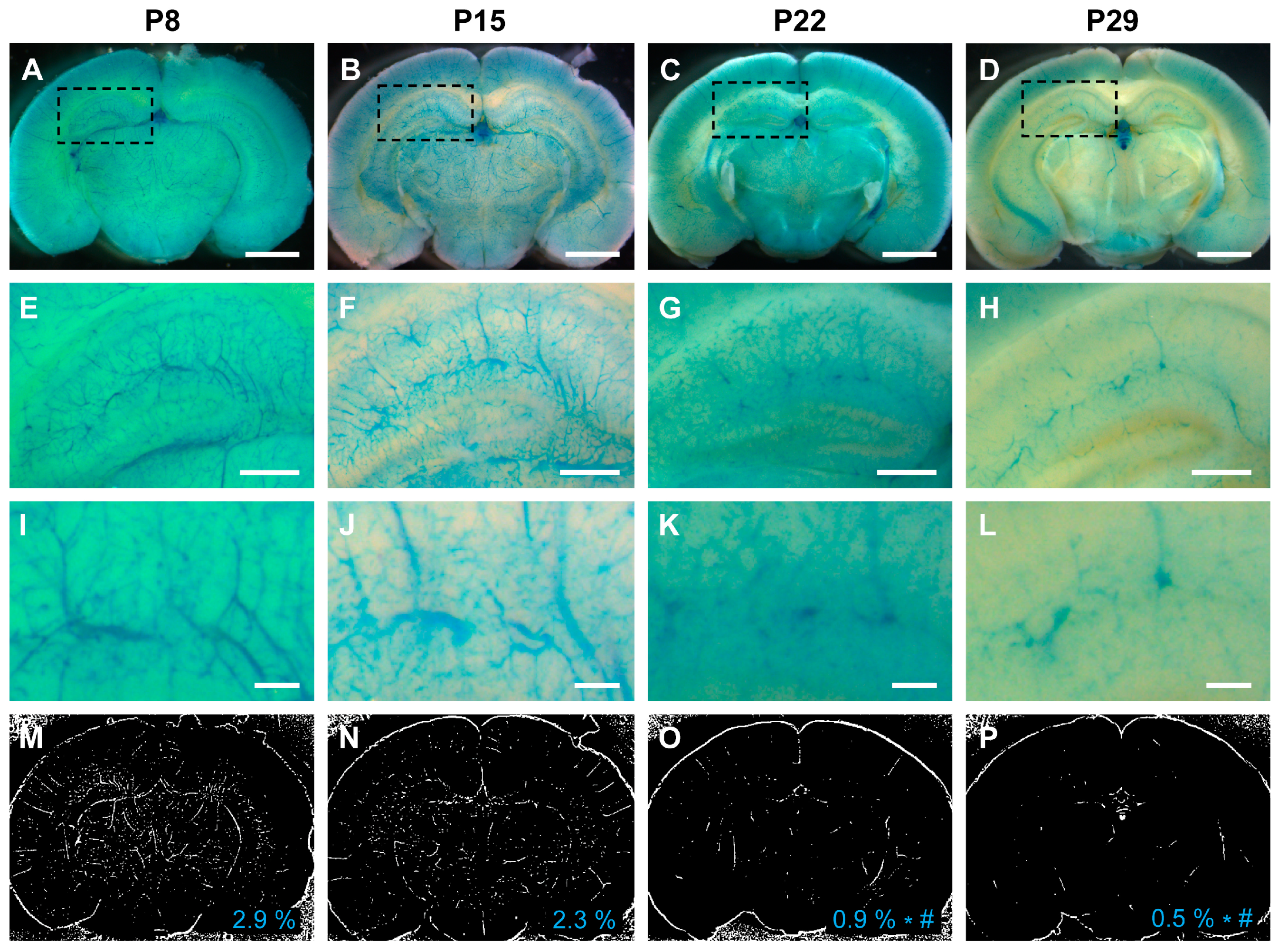
3.4. VEGFR2 Expression Predominated in the Early Postnatal Period
4. Discussion
4.1. Postnatal Development of BAVMs in a Type I HHT Mouse Model
4.2. The Congenital Nature of BAVMs
4.3. Angiogenesis as an Endogenous Tertiary Hit after Genetic Second Hit
4.4. Pathogenesis of De Novo BAVMs
4.5. A Novel Mouse Model for Forebrain and Hindbrain BAVMs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Florian, I.A.; Beni, L.; Moisoiu, V.; Timis, T.L.; Florian, I.S.; Balasa, A.; Berindan-Neagoe, I. ‘De Novo’ Brain AVMs-Hypotheses for Development and a Systematic Review of Reported Cases. Medicina 2021, 57, 201. [Google Scholar] [CrossRef] [PubMed]
- Pabaney, A.H.; Rammo, R.A.; Tahir, R.A.; Seyfried, D. Development of De Novo Arteriovenous Malformation Following Ischemic Stroke: Case Report and Review of Current Literature. World Neurosurg. 2016, 96, e608.e5–e608.e12. [Google Scholar] [CrossRef]
- Dogan, S.N.; Bagcilar, O.; Mammadov, T.; Kizilkilic, O.; Islak, C.; Kocer, N. De Novo Development of a Cerebral Arteriovenous Malformation: Case Report and Review of the Literature. World Neurosurg. 2019, 126, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quinones, C.; Koch, M.J.; Raymond, S.B.; Patel, A. Left Thalamus Arteriovenous Malformation Secondary to Radiation Therapy of Original Vermian Arteriovenous Malformation: Case Report. J. Stroke Cerebrovasc. Dis. 2019, 28, e53–e59. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, A.; Rogers, J.M.; Assaad, N.N.A.; Rodriguez, M.L.; Stoodley, M.A.; Morgan, M.K. De novo brain arteriovenous malformation after tumor resection: Case report and literature review. Acta Neurochir. 2018, 160, 2191–2197. [Google Scholar] [CrossRef]
- Rodrigues de Oliveira, L.F.; Castro-Afonso, L.H.; Freitas, R.K.; Colli, B.O.; Abud, D.G. De Novo Intracranial Arteriovenous Malformation-Case Report and Literature Review. World Neurosurg. 2020, 138, 349–351. [Google Scholar] [CrossRef]
- Gonzalez, L.F.; Bristol, R.E.; Porter, R.W.; Spetzler, R.F. De novo presentation of an arteriovenous malformation. Case report and review of the literature. J. Neurosurg. 2005, 102, 726–729. [Google Scholar] [CrossRef]
- Shimoda, Y.; Osanai, T.; Nakayama, N.; Ushikoshi, S.; Hokari, M.; Shichinohe, H.; Abumiya, T.; Kazumata, K.; Houkin, K. De novo arteriovenous malformation in a patient with hereditary hemorrhagic telangiectasia. J. Neurosurg. Pediatr. 2016, 17, 330–335. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef]
- Hong, T.; Yan, Y.; Li, J.; Radovanovic, I.; Ma, X.; Shao, Y.W.; Yu, J.; Ma, Y.; Zhang, P.; Ling, F.; et al. High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain 2019, 142, 23–34. [Google Scholar] [CrossRef]
- Oka, M.; Kushamae, M.; Aoki, T.; Yamaguchi, T.; Kitazato, K.; Abekura, Y.; Kawamata, T.; Mizutani, T.; Miyamoto, S.; Takagi, Y. KRAS G12D or G12V Mutation in Human Brain Arteriovenous Malformations. World Neurosurg. 2019, 126, e1365–e1373. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Dumont, D.J.; Letarte, M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 1999, 104, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular deficiency of Smad4 causes arteriovenous malformations: A mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Park, S.O.; Wankhede, M.; Lee, Y.J.; Choi, E.J.; Fliess, N.; Choe, S.W.; Oh, S.H.; Walter, G.; Raizada, M.K.; Sorg, B.S.; et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 2009, 119, 3487–3496. [Google Scholar] [CrossRef]
- Satomi, J.; Mount, R.J.; Toporsian, M.; Paterson, A.D.; Wallace, M.C.; Harrison, R.V.; Letarte, M. Cerebral vascular abnormalities in a murine model of hereditary hemorrhagic telangiectasia. Stroke 2003, 34, 783–789. [Google Scholar] [CrossRef]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Nguyen, H.L.; Cunningham, T.A.; Choe, S.W.; Jiang, Z.; Arthur, H.M.; Lee, Y.J.; Oh, S.P. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2232–2236. [Google Scholar] [CrossRef]
- Choi, E.J.; Walker, E.J.; Shen, F.; Oh, S.P.; Arthur, H.M.; Young, W.L.; Su, H. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 2012, 33, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Chen, W.; Jun, K.; Arthur, H.M.; Young, W.L.; Su, H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS ONE 2014, 9, e88511. [Google Scholar] [CrossRef] [PubMed]
- Scherschinski, L.; Han, C.; Kim, Y.H.; Winkler, E.A.; Catapano, J.S.; Schriber, T.D.; Vajkoczy, P.; Lawton, M.T.; Oh, S.P. Localized conditional induction of brain arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Angiogenesis 2023, 26, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Milton, I.; Ouyang, D.; Allen, C.J.; Yanasak, N.E.; Gossage, J.R.; Alleyne, C.H., Jr.; Seki, T. Age-dependent lethality in novel transgenic mouse models of central nervous system arteriovenous malformations. Stroke 2012, 43, 1432–1435. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Garcia-Collado, M.; Redgrave, R.E.; Singh, E.; Davison, B.; Park, C.; Lin, H.; Luli, S.; Jin, Y.; Wang, Y.; et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ. Res. 2020, 126, 243–257. [Google Scholar] [CrossRef]
- Walker, E.J.; Su, H.; Shen, F.; Degos, V.; Amend, G.; Jun, K.; Young, W.L. Bevacizumab attenuates VEGF-induced angiogenesis and vascular malformations in the adult mouse brain. Stroke 2012, 43, 1925–1930. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, S.; Huang, S.; Yoo, J.Y.; Korbelin, J.; Lee, T.J.; Kaur, B.; Dash, P.K.; Chen, P.R.; Kim, E. Selective Endothelial Hyperactivation of Oncogenic KRAS Induces Brain Arteriovenous Malformations in Mice. Ann. Neurol. 2021, 89, 926–941. [Google Scholar] [CrossRef]
- Fish, J.E.; Flores Suarez, C.P.; Boudreau, E.; Herman, A.M.; Gutierrez, M.C.; Gustafson, D.; DiStefano, P.V.; Cui, M.; Chen, Z.; De Ruiz, K.B.; et al. Somatic Gain of KRAS Function in the Endothelium Is Sufficient to Cause Vascular Malformations That Require MEK but Not PI3K Signaling. Circ. Res. 2020, 127, 727–743. [Google Scholar] [CrossRef]
- Allinson, K.R.; Carvalho, R.L.; van den Brink, S.; Mummery, C.L.; Arthur, H.M. Generation of a floxed allele of the mouse Endoglin gene. Genesis 2007, 45, 391–395. [Google Scholar] [CrossRef]
- Gothert, J.R.; Gustin, S.E.; van Eekelen, J.A.; Schmidt, U.; Hall, M.A.; Jane, S.M.; Green, A.R.; Gottgens, B.; Izon, D.J.; Begley, C.G. Genetically tagging endothelial cells in vivo: Bone marrow-derived cells do not contribute to tumor endothelium. Blood 2004, 104, 1769–1777. [Google Scholar] [CrossRef]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Choe, S.W.; Kim, Y.H.; Acharya, A.P.; Keselowsky, B.G.; Sorg, B.S.; Lee, Y.J.; Oh, S.P. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 2014, 17, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Lang, M.J.; Nguyen, C.L.; Luna Melendez, E.; Mehta, S.; Turner, G.H.; Lawton, M.T.; Oh, S.P. Novel experimental model of brain arteriovenous malformations using conditional Alk1 gene deletion in transgenic mice. J. Neurosurg. 2021, 137, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Han, Z.; Jun, K.; Camus, M.; Wankhede, M.; Mao, L.; Arnold, T.; Young, W.L.; Su, H. De novo cerebrovascular malformation in the adult mouse after endothelial Alk1 deletion and angiogenic stimulation. Stroke 2014, 45, 900–902. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Garel, S. On place and time: Microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 2017, 47, 121–130. [Google Scholar] [CrossRef]
- Robertson, P.L.; Du Bois, M.; Bowman, P.D.; Goldstein, G.W. Angiogenesis in developing rat brain: An in vivo and in vitro study. Brain Res. 1985, 355, 219–223. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Kremer, C.; Breier, G.; Risau, W.; Plate, K.H. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997, 57, 3852–3859. [Google Scholar]
- Abdalla, S.A.; Pece-Barbara, N.; Vera, S.; Tapia, E.; Paez, E.; Bernabeu, C.; Letarte, M. Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum. Mol. Genet. 2000, 9, 1227–1237. [Google Scholar] [CrossRef]
- Pece, N.; Vera, S.; Cymerman, U.; White, R.I., Jr.; Wrana, J.L.; Letarte, M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J. Clin. Investig. 1997, 100, 2568–2579. [Google Scholar] [CrossRef]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am. J. Hum. Genet. 2019, 105, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Ali, M.K.; Tan, S.Y.; Teng, J.; Do, H.M.; Steinberg, G.K.; Stevenson, D.A.; Spiekerkoetter, E. Arteriovenous Malformations-Current Understanding of the Pathogenesis with Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 9037. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Santos, V.; Shih, A.Y. Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e363. [Google Scholar] [CrossRef]
- Harb, R.; Whiteus, C.; Freitas, C.; Grutzendler, J. In vivo imaging of cerebral microvascular plasticity from birth to death. J. Cereb. Blood Flow. Metab. 2013, 33, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Walchli, T.; Mateos, J.M.; Weinman, O.; Babic, D.; Regli, L.; Hoerstrup, S.P.; Gerhardt, H.; Schwab, M.E.; Vogel, J. Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat. Protoc. 2015, 10, 53–74. [Google Scholar] [CrossRef]
- Zeller, K.; Vogel, J.; Kuschinsky, W. Postnatal distribution of Glut1 glucose transporter and relative capillary density in blood-brain barrier structures and circumventricular organs during development. Brain Res. Dev. Brain Res. 1996, 91, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Vates, G.E.; Hashimoto, T.; Young, W.L.; Lawton, M.T. Angiogenesis in the brain during development: The effects of vascular endothelial growth factor and angiopoietin-2 in an animal model. J. Neurosurg. 2005, 103, 136–145. [Google Scholar] [CrossRef]
- Brinjikji, W.; Iyer, V.N.; Yamaki, V.; Lanzino, G.; Cloft, H.J.; Thielen, K.R.; Swanson, K.L.; Wood, C.P. Neurovascular Manifestations of Hereditary Hemorrhagic Telangiectasia: A Consecutive Series of 376 Patients during 15 Years. AJNR Am. J. Neuroradiol. 2016, 37, 1479–1486. [Google Scholar] [CrossRef]
- Lunsford, L.D.; Niranjan, A.; Kondziolka, D.; Sirin, S.; Flickinger, J.C. Arteriovenous malformation radiosurgery: A twenty year perspective. Clin. Neurosurg. 2008, 55, 108–119. [Google Scholar] [PubMed]
- Yang, W.Y.; Luo, C.B.; Tsuei, Y.S.; Guo, W.Y.; Wu, H.M.; Chung, W.Y. A single-institution study of predisposing factors of patients with BAVMs to flow-related aneurysm. J. Formos. Med. Assoc. 2019, 118, 707–712. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Nguyen, C.L.; Scherschinski, L.; Schriber, T.D.; Arthur, H.M.; Lawton, M.T.; Oh, S.P. VEGFR2 Expression Correlates with Postnatal Development of Brain Arteriovenous Malformations in a Mouse Model of Type I Hereditary Hemorrhagic Telangiectasia. Biomedicines 2023, 11, 3153. https://doi.org/10.3390/biomedicines11123153
Han C, Nguyen CL, Scherschinski L, Schriber TD, Arthur HM, Lawton MT, Oh SP. VEGFR2 Expression Correlates with Postnatal Development of Brain Arteriovenous Malformations in a Mouse Model of Type I Hereditary Hemorrhagic Telangiectasia. Biomedicines. 2023; 11(12):3153. https://doi.org/10.3390/biomedicines11123153
Chicago/Turabian StyleHan, Chul, Candice L. Nguyen, Lea Scherschinski, Tyler D. Schriber, Helen M. Arthur, Michael T. Lawton, and Suk Paul Oh. 2023. "VEGFR2 Expression Correlates with Postnatal Development of Brain Arteriovenous Malformations in a Mouse Model of Type I Hereditary Hemorrhagic Telangiectasia" Biomedicines 11, no. 12: 3153. https://doi.org/10.3390/biomedicines11123153
APA StyleHan, C., Nguyen, C. L., Scherschinski, L., Schriber, T. D., Arthur, H. M., Lawton, M. T., & Oh, S. P. (2023). VEGFR2 Expression Correlates with Postnatal Development of Brain Arteriovenous Malformations in a Mouse Model of Type I Hereditary Hemorrhagic Telangiectasia. Biomedicines, 11(12), 3153. https://doi.org/10.3390/biomedicines11123153