
Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Transporter-Mediated Interactions between Glycine and Glutamate
1.2. The Possible Gly–Glu Cotransmission
1.3. Aims of the Study and Main Conclusions
2. Materials and Methods
2.1. Animals
2.2. Preparation of Synaptosomes
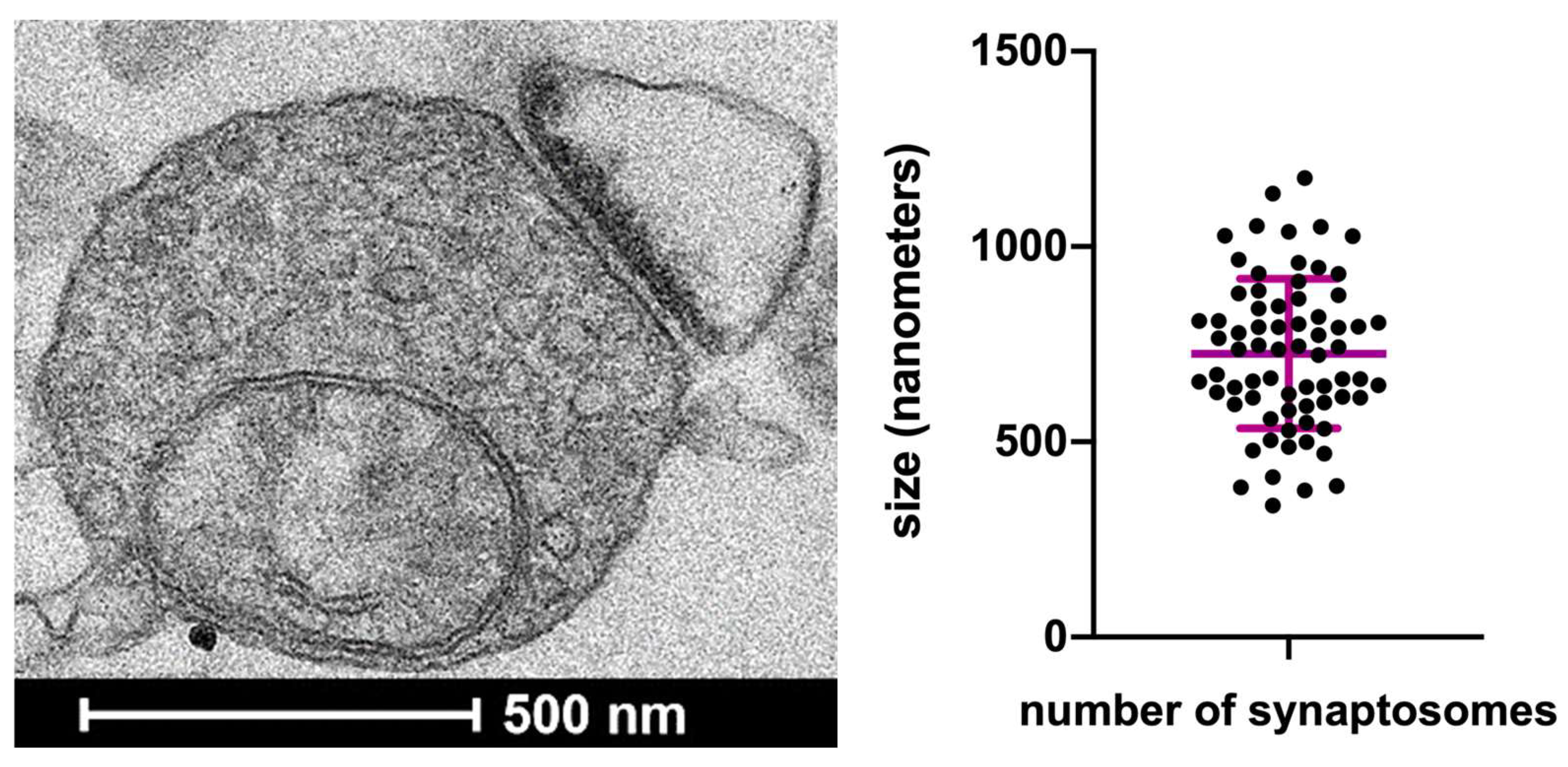
2.3. Electron Microscopy
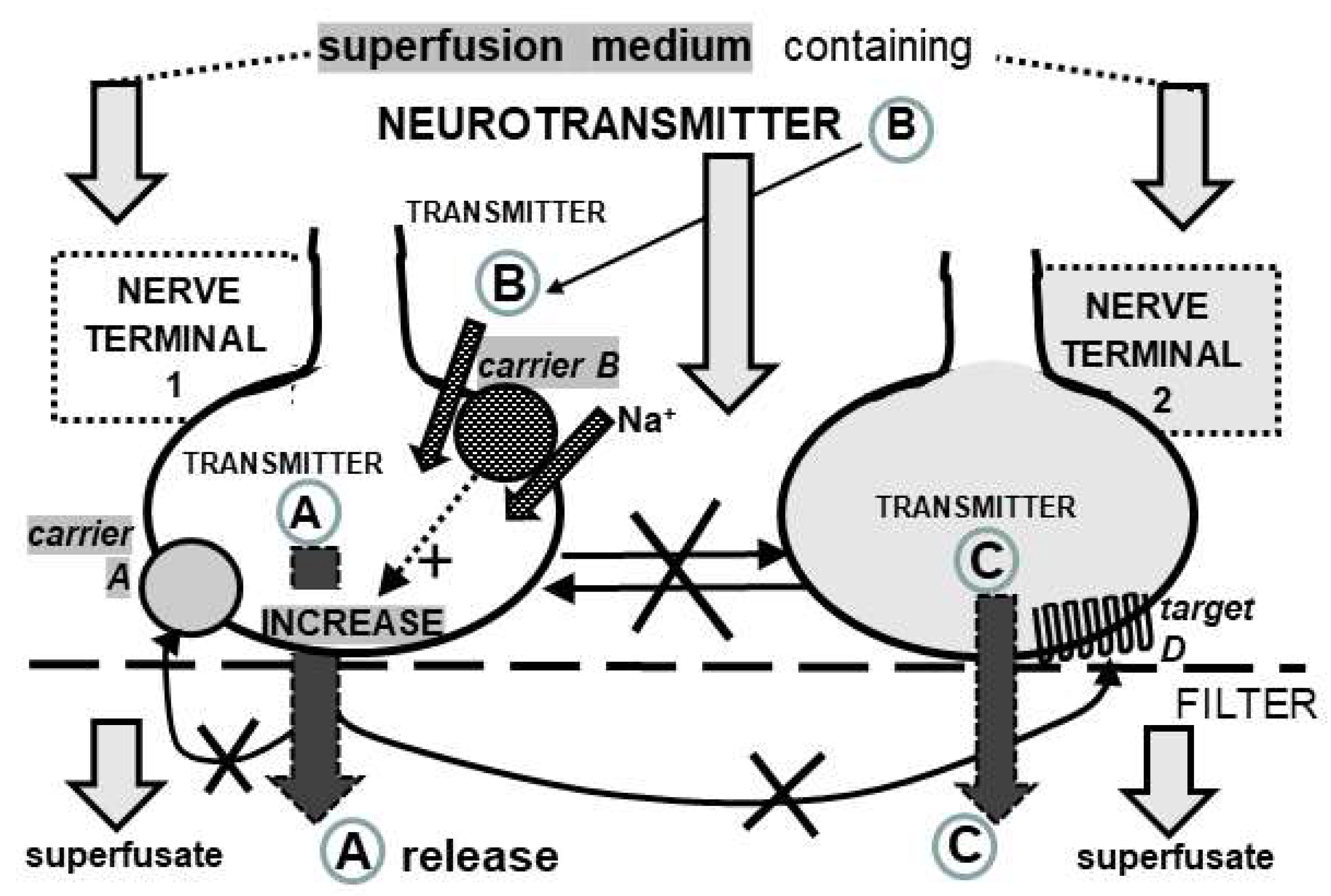
2.4. Neurotransmitter Release Experiments
2.5. Data Analysis for Release Experiments
2.6. Chemicals
3. Results
3.1. The Experimental Model: Ultrastructural Analysis
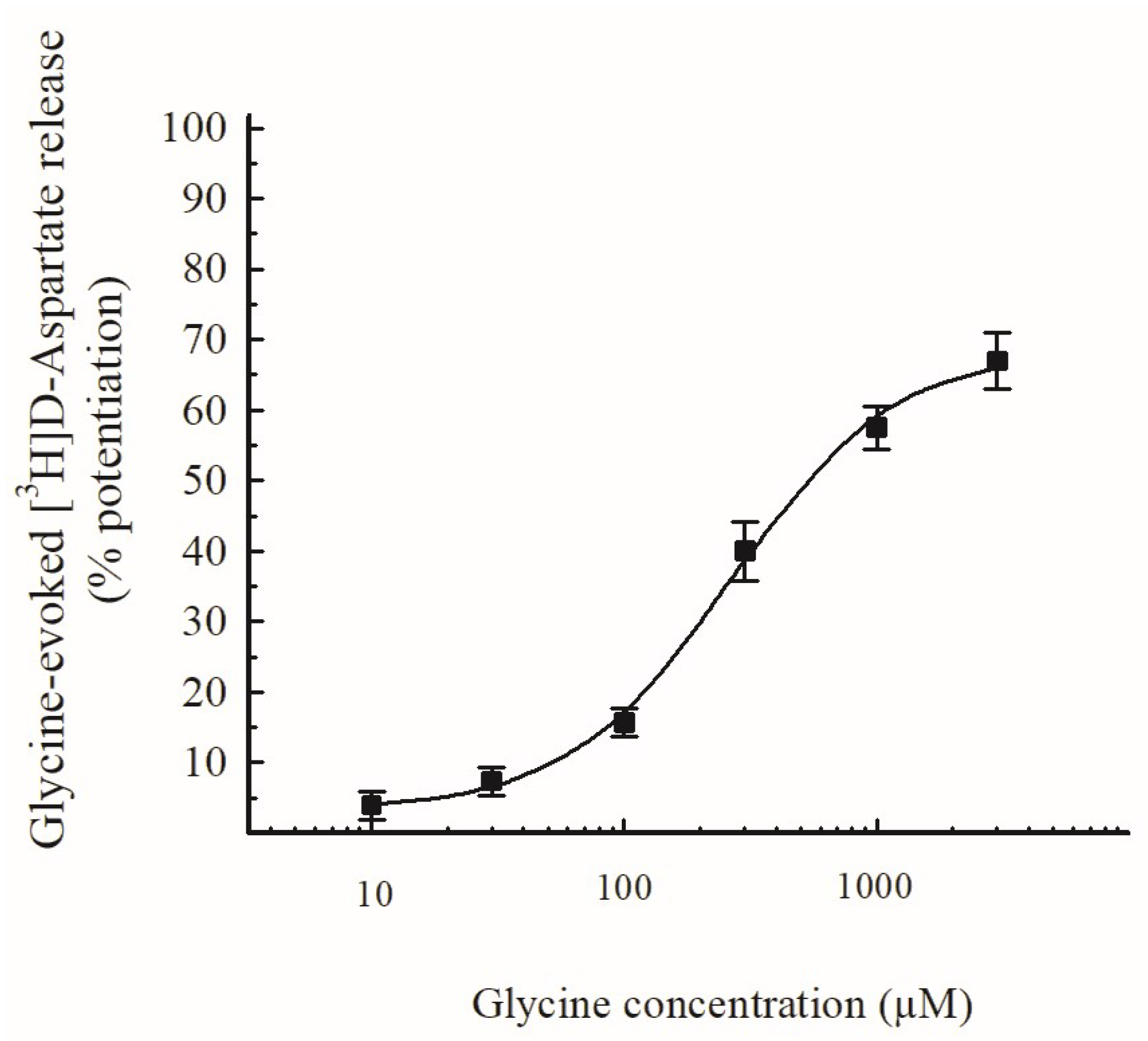
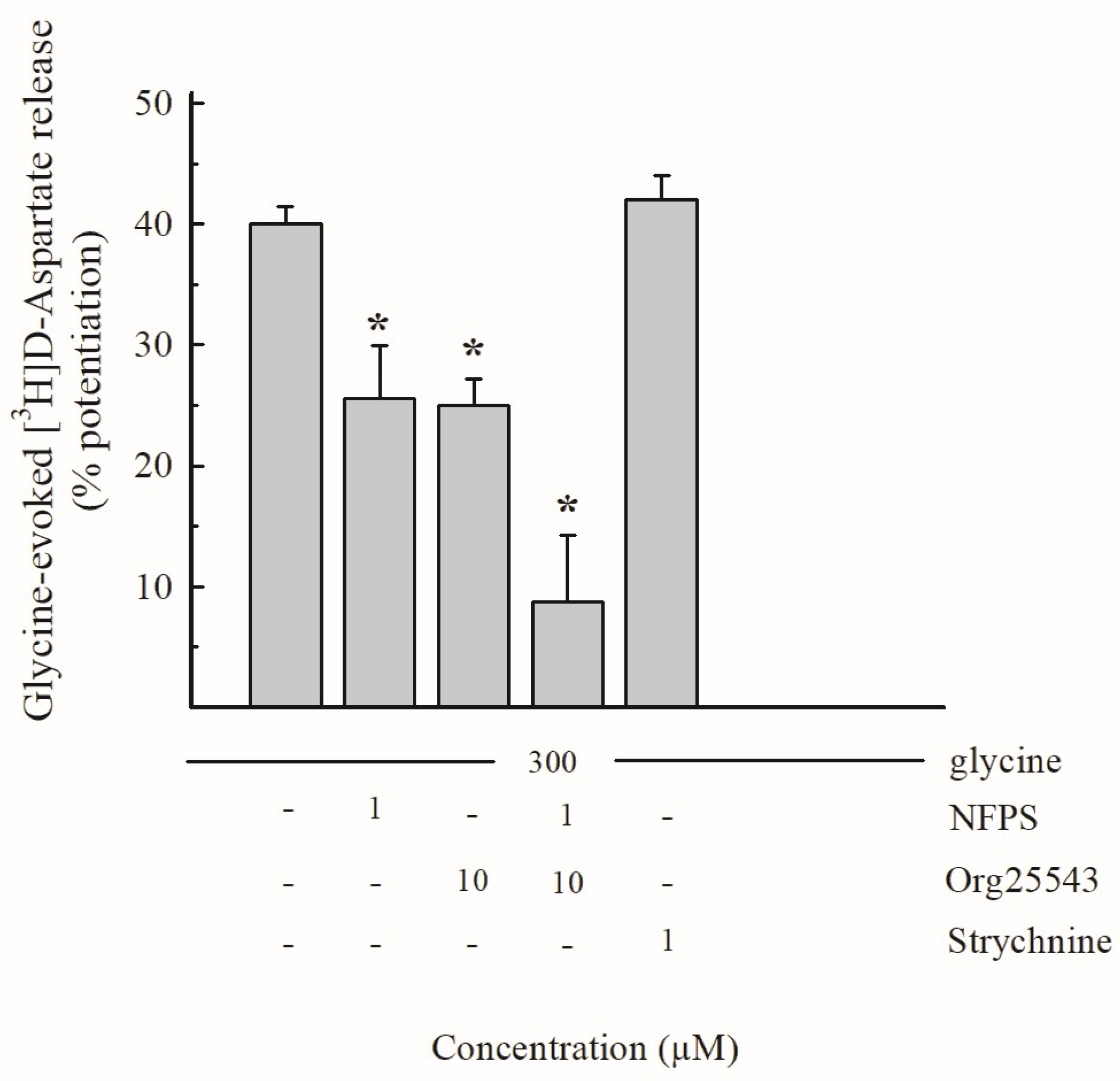
3.2. Effects of Gly and of Selective GlyT Blockers in Hippocampal Glutamate-Releasing Nerve Terminals
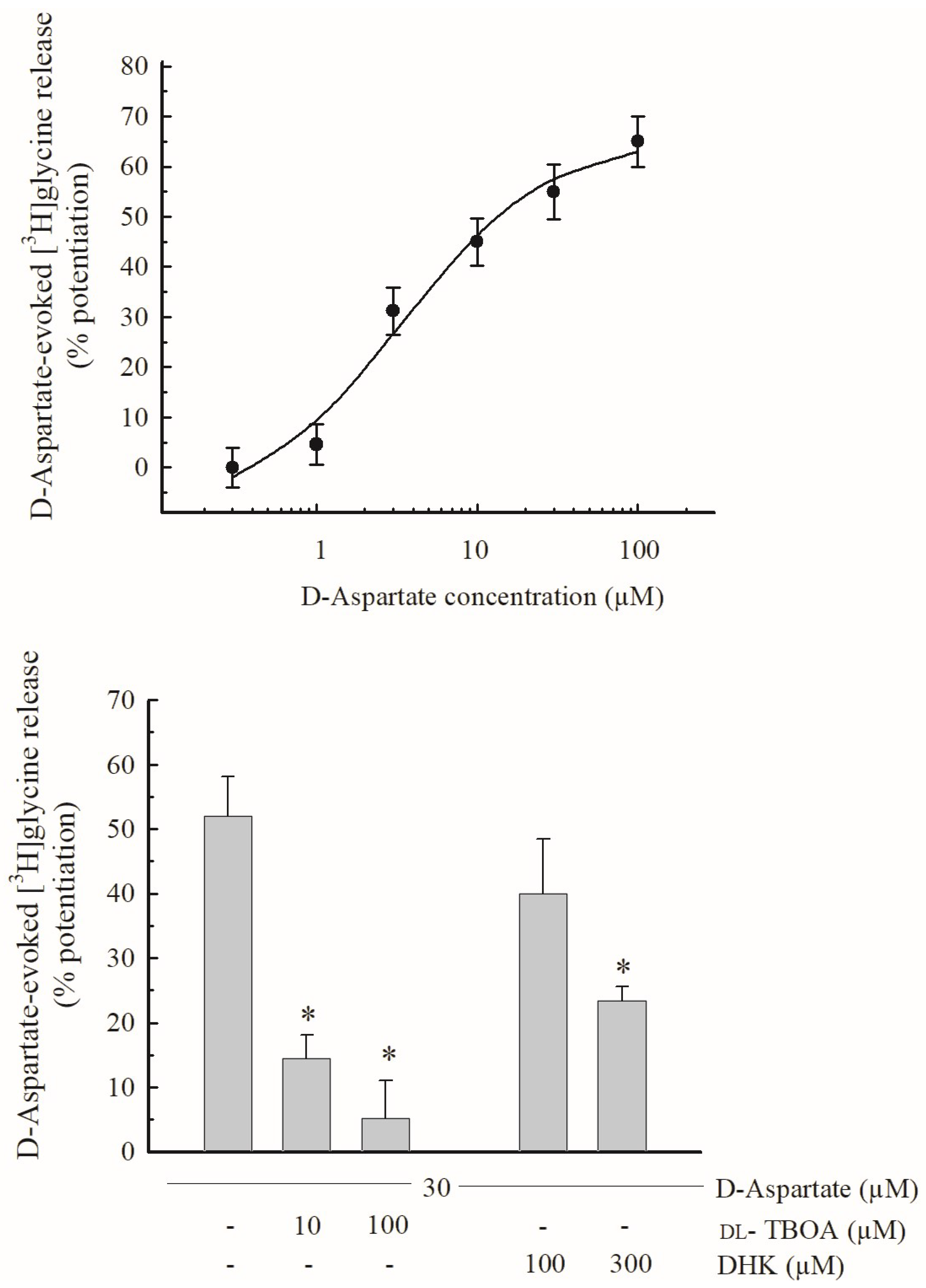
3.3. Effects of D-Asp and of EAAT Blockers on the Release of [3H]glycine
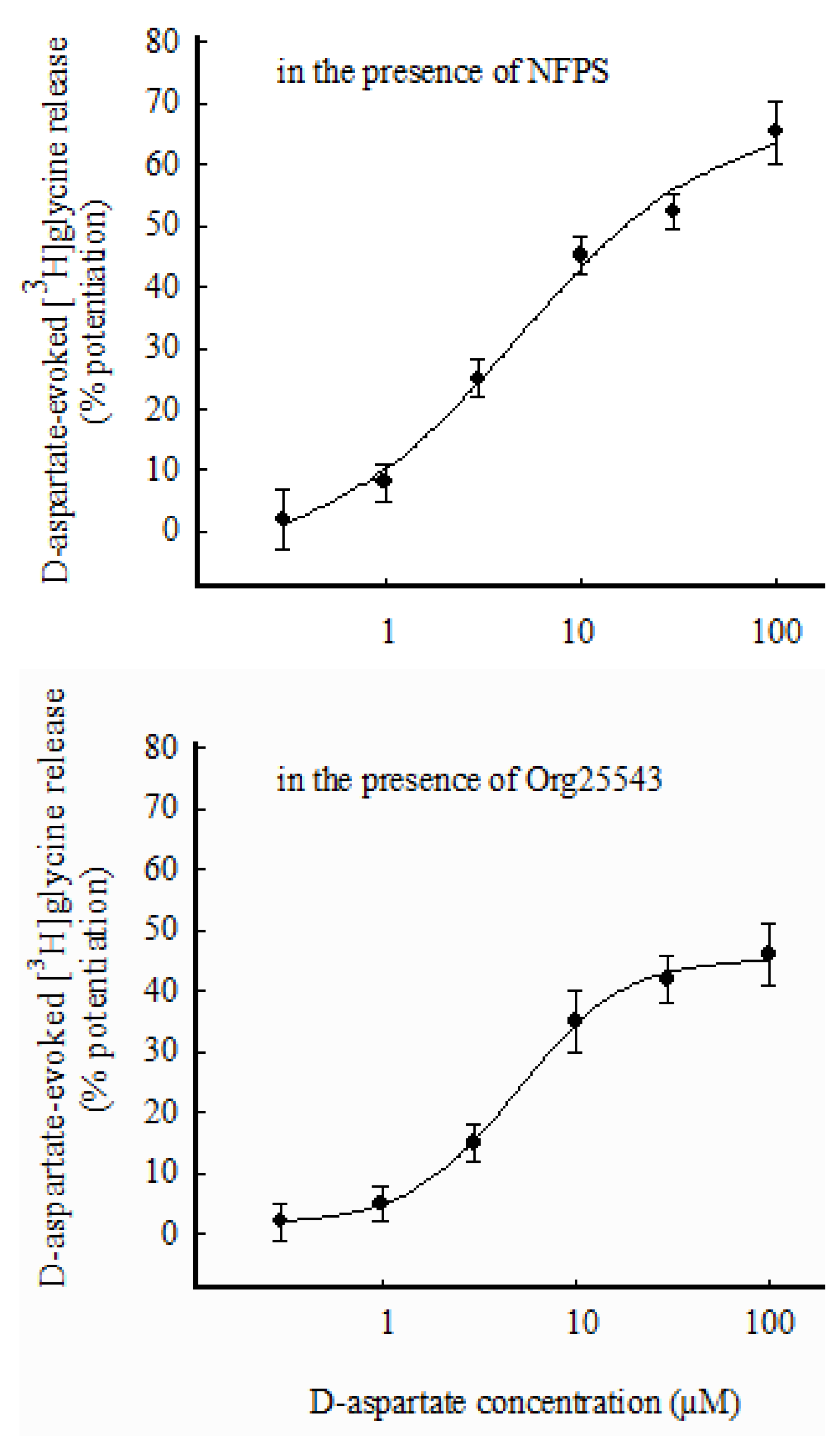
3.4. Selective Labeling through GlyT1 or GlyT2 and Effects of D-Asp on [3H]glycine Release
4. Discussion
4.1. Considerations on the Experimental Model and Technique
4.2. Localization and Functions of GlyTs on Hippocampal Glutamate-Releasing Nerve Terminals
4.3. Considerations on the GlyT Types Found on Mouse Hippocampal Glu-Releasing Nerve Terminals
- (i)
- It was proposed that even a few GlyT2 transporters, barely detectable through morphological approaches, can accumulate enough Gly that it can be measured by functional uptake assays [80], possibly also due to the very efficient accumulative power associated with GlyT2-mediated uptake: as established by Roux and Supplisson [85], GlyT1 has a stoichiometry of 2Na+/Cl−/Gly, while the stoichiometry of GlyT2 was reported to be 3 Na+/Cl−/Gly, so that the driving force for Gly uphill transport is much larger with GlyT2 than GlyT1 [86]. Similarly, we suggest that a few GlyT2 on a possibly small subset of hippocampal nerve terminals, able to release [3H]D-Asp/Glu, are activated by Gly due to their strong accumulative power with enough efficiency to elicit a measurable functional response in experiments in which the release of preloaded [3H]D-Asp induced by Gly is monitored (present work).
- (ii)
- According to the considerations above, the synaptosomal subpopulation considered here could represent a very low percentage of the entire population of nerve terminals; in these conditions, both an advantage and a caveat of this preparation become evident: advantages of synaptosomes include the possibility of obtaining functional results even when the targets under study are poorly expressed in a certain CNS area [23,58]. While it is expected that functional results should be confirmed by immunochemical data that show the presence of the target structures under study, unfortunately, due to the reasons just discussed, this might be hard or impossible if such targets (for example, GlyT2 on Glu-releasing terminals) are poorly expressed (see [58], p. 1003).
4.4. Localization and Functions of EAATs on Hippocampal Nerve Terminals That Release [3H]glycine and Considerations of Possible Involvement of Other Glutamatergic Targets
4.5. Selective Labeling through GlyT1 or GlyT2 Does Not Permit Us to Establish GlyT1/GlyT2 Coexistence or Segregation on EAAT-Bearing Nerve Terminals
4.6. Considerations on Possible Modes of Interaction between Gly and Glu
4.7. Considerations on Gly–Glu Possible Cotransmission
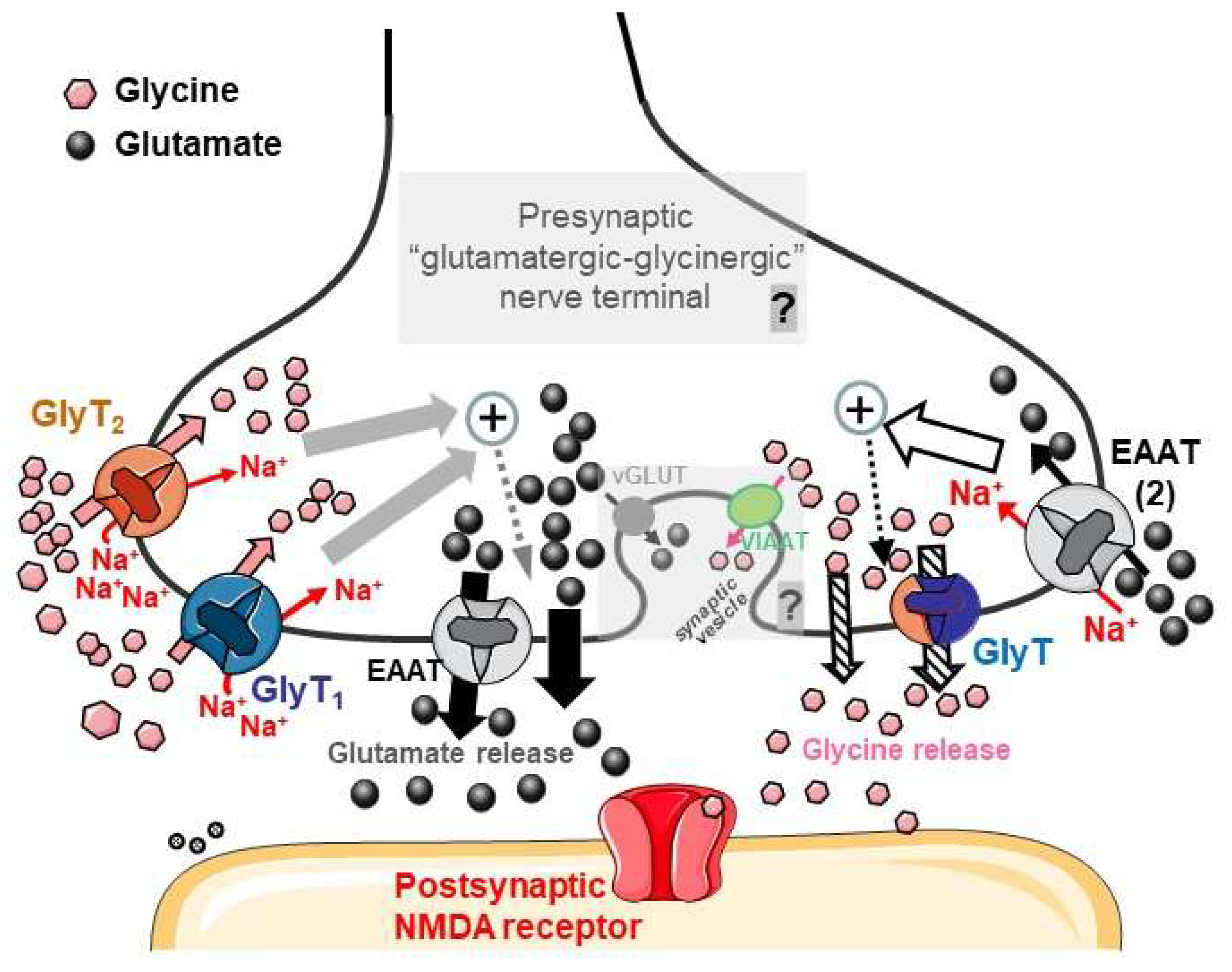
- Gly, either released by the “glutamatergic-glycinergic nerve terminals” proposed here or originating from other sources (neighboring neurons, glial cells, metabolic sources…), is a substrate of the Na+ -dependent GlyT1 and GlyT2 transporters, which can deliver Gly to the intraterminal space; thus, the amino acid can be stored in the same terminal with Glu. The Na+/Gly cotransport triggers internal events leading to an increased release of Glutamate (gray and gray-dotted arrows) through mechanisms including the facilitation of EAAT reversal and other, likely, non-exocytotic mechanisms (thick black arrows; see Section 4.6).
- D-Aspartate or endogenous Glutamate activates EAATs (including transporters of the EAAT2 type) that are Na+-dependent. The cotransport of the excitatory amino acid and Na+ triggers presynaptic events (white arrow and black dotted arrow), leading to an increase in Gly release that can occur through mechanisms including, possibly, the reversal of GlyTs (“crossed out” arrows).
- The most speculative concepts illustrated in the figure are pointed out in gray, alongside a “question mark”. The possible Gly–Glu cotransmission would be compatible with the costorage of Gly and Glu in synaptic vesicles through VIAAT-VGAT and vGluT vesicular transporters, respectively (see Section 4.7). The cotransmitters could then be coreleased as coagonists onto postsynaptic NMDA receptors. A final demonstration of these concepts (presented in the “C” paragraph) will require further investigation.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.; Meyer, T.M.; Coyle, J.T.; Greene, R.W. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc. Natl. Acad. Sci. USA 1998, 95, 15730–15734. [Google Scholar] [CrossRef]
- Labrie, V.; Lipina, T.; Roder, J.C. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology 2008, 200, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov. 2013, 12, 866–885. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Bakkar, W.; Martina, M.; Sokolovski, A.; Wong, A.; Legendre, P.; Bergeron, R. Vesicular storage of glycine in glutamatergic terminals in mouse hippocampus. Neuroscience 2013, 242, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Gimenez, C.; Zafra, F. Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb. Cortex 2005, 15, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Leite, C.; Giménez, C.; Zafra, F. Localization of the glycine transporter GLYT1 in glutamatergic synaptic vesicles. Neurochem. Int. 2014, 73, 204–210. [Google Scholar] [CrossRef]
- Cioffi, C.L.; Guzzo, P.R. Inhibitors of glycine transporter-1: Potential therapeutics for the treatment of CNS disorders. Curr. Top. Med. Chem. 2016, 16, 3404–3437. [Google Scholar] [CrossRef]
- Piniella, D.; Zafra, F. Functional crosstalk of the glycine transporter GlyT1 and NMDA receptors. Neuropharmacology 2023, 232, 109514. [Google Scholar] [CrossRef]
- Starke, K.; Gothert, M.; Kilbinger, H.; Pinho, D.; Quintas, C.; Sardo, F.; Cardoso, T.M.; Queiroz, G.; Behn, C.G.D.; Booth, V.; et al. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989, 69, 864–989. [Google Scholar] [CrossRef]
- Langer, S.Z. Therapeutic use of release-modifying drugs. Handb. Exp. Pharmacol. 2008, 184, 561–573. [Google Scholar] [CrossRef]
- Raiteri, M. Presynaptic metabotropic glutamate and GABAB receptors. Handb. Exp. Pharmacol. 2008, 184, 373–407. [Google Scholar] [CrossRef]
- Feuerstein, T.J. Presynaptic receptors for dopamine, histamine, and serotonin. Handb. Exp. Pharmacol. 2008, 184, 289–338. [Google Scholar] [CrossRef]
- Bonanno, G.; Raiteri, M. Release-regulating presynaptic heterocarriers. Prog. Neurobiol. 1994, 44, 451–462. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M.; Bonanno, G. Coexistence and function of different neurotransmitter transporters in the plasma membrane of CNS neurons. Prog. Neurobiol. 2002, 68, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Raiteri, L.; Paluzzi, S.; Zappettini, S.; Usai, C.; Raiteri, M. Co-Existence of GABA and Glu Transporters in the Central Nervous System. Curr. Top. Med. Chem. 2006, 6, 979–988. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Synaptosomes Still Viable after 25 Years of Superfusion. Neurochem. Res. 2000, 25, 1265–1274. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Siri, A.; Passalacqua, M.; Melloni, E.; Raiteri, M.; Bonanno, G. Glycine taken up through GLYT1 and GLYT2 heterotransporters into glutamatergic axon terminals of mouse spinal cord elicits release of glutamate by homotransporter reversal and through anion channels. Biochem. Pharmacol. 2005, 69, 159–168. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Patti, L.; Usai, C.; Bucci, G.; Diaspro, A.; Raiteri, M.; Bonanno, G. Activation of gamma-aminobutyric acid GAT-1 transporters on glutamatergic terminals of mouse spinal cord mediates glutamate release through anion channels and by transporter reversal. J. Neurosci. Res. 2005, 80, 424–433. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Usai, C.; Diaspro, A.; Paluzzi, S.; Milanese, M.; Raiteri, M.; Bonanno, G. Functional expression of release-regulating glycine transporters GLYT1 on GABAergic neurons and GLYT2 on astrocytes in mouse spinal cord. Neurochem. Int. 2008, 52, 103–112. [Google Scholar] [CrossRef]
- Milanese, M.; Romei, C.; Usai, C.; Oliveri, M.; Raiteri, L. A new function for glycine GlyT2 transporters: Stimulation of γ-aminobutyric acid release from cerebellar nerve terminals through GAT1 transporter reversal and Ca(2+)-dependent anion channels. J. Neurosci. Res. 2014, 92, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis regulates trafficking of GABA and glycine heterotransporters in spinal cord glutamatergic synapses: A mechanism for the excessive heterotransporter-induced release of glutamate in experimental amyotrophic lateral sclerosis. Neurobiol. Dis. 2015, 74, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Bonifacino, T.; Milanese, M.; Usai, C.; Raiteri, L. Colocalization of neurotransmitter transporters on the plasma membrane of the same nerve terminal may reflect cotransmission. Brain Res. Bull. 2016, 127, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Vallebuona, F.; Donadini, F.; Fontana, G.; Fedele, E.; Raiteri, M. Heterocarrier-mediated reciprocal modulation of glutamate and glycine release in rat cerebral cortex and spinal cord synaptosomes. Eur. J. Pharmacol. 1994, 252, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Paolucci, E.; Prisco, S.; Raiteri, M.; Bonanno, G. Activation of a glycine transporter on spinal cord neurons causes enhanced glutamate release in a mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2003, 138, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Stigliani, S.; Zappettini, S.; Mercuri, N.B.; Raiteri, M.; Bonanno, G. Excessive and precocious glutamate release in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 2004, 46, 782–792. [Google Scholar] [CrossRef]
- Waseem, T.V.; Fedorovich, S.V. Presynaptic glycine receptors influence plasma membrane potential and glutamate release. Neurochem. Res. 2010, 35, 1188–1195. [Google Scholar] [CrossRef]
- Musante, V.; Summa, M.; Cunha, R.A.; Raiteri, M.; Pittaluga, A. Pre-synaptic glycine GlyT1 transporter—NMDA receptor interaction: Relevance to NMDA autoreceptor activation in the presence of Mg2+ ions. J. Neurochem. 2011, 117, 516–527. [Google Scholar] [CrossRef]
- Harsing, L.G., Jr.; Matyus, P. Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: The role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 2013, 93, 110–119. [Google Scholar] [CrossRef]
- Marshak, D.W.; Chuang, A.Z.; Dolino, D.M.; Jacoby, R.A.; Liu, W.S.; Long, Y.; Sherman, M.B.; Suh, J.M.; Vila, A.; Mills, S.L. Synaptic connections of amacrine cells containing vesicular glutamate transporter 3 in baboon retinas. Vis. Neurosci. 2015, 32, E006. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, Y.; Chen, M.; Zhou, Z.J. Segregated Glycine-Glutamate Co-transmission from vGluT3 Amacrine Cells to Contrast-Suppressed and Contrast-Enhanced Retinal Circuits. Neuron 2016, 90, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, F.; Morales, M.A. Functional Implications of Neurotransmitter Segregation. Front. Neural Circuits 2021, 15, 738516. [Google Scholar] [CrossRef] [PubMed]
- Marques, B.L.; Oliveira-Lima, O.C.; Carvalho, G.A.; de Almeida Chiarelli, R.; Ribeiro, R.I.; Parreira, R.C.; da Madeira Freitas, E.M.; Resende, R.R.; Klempin, F.; Ulrich, H.; et al. Neurobiology of glycine transporters: From molecules to behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Cotransmission. Curr. Opin. Pharmacol. 2003, 4, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Apergis-Schoute, J.; Burnstock, G.; Nusbaum, M.P.; Parker, D.; Schiöth, H.B. General Principles of Neuronal Co-transmission: Insights from Multiple Model Systems. Front. Neural Circuits 2019, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Bischofberger, J.; Sandkühler, J. Corelease of two fast neurotransmitters at a central synapse. Science 1998, 281, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, A.; Triller, A.; Dieudonné, S. IPSC Kinetics at Identified GABAergic and Mixed GABAergic and Glycinergic Synapses onto Cerebellar Golgi Cells. J. Neurosci. 2001, 21, 6045–6057. [Google Scholar] [CrossRef]
- Simat, M.; Parpan, F.; Fritschy, J. Heterogeneity of glycinergic and gabaergic interneurons in the granule cell layer of mouse cerebellum. J. Comp. Neurol. 2007, 500, 71–83. [Google Scholar] [CrossRef]
- Romei, C.; Raiteri, M.; Raiteri, L. GABA transporters mediate glycine release from cerebellum nerve endings: Roles of Ca2+ channels, mitochondrial Na+/Ca2+ exchangers, vesicular GABA/glycine transporters and anion channels. Neurochem. Int. 2012, 61, 133–140. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Heath, J.W.; Harrison, S.M.; Jarvie, P.E.; Glenfield, P.J.; Rostas, J.A. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: Homogeneity and morphology of subcellular fractions. Brain Res. 1988, 441, 59–71. [Google Scholar] [CrossRef]
- Nakamura, Y.; Iga, K.; Shibata, T.; Shudo, M.; Kataoka, K. Glial plasmalemmal vesicles: A subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia 1993, 9, 48–56. [Google Scholar] [CrossRef]
- Luccini, E.; Raiteri, L. Mechanisms of [3H]glycine release from mouse spinal cord synaptosomes selectively labeled through GLYT2 transporters. J. Neurochem. 2007, 103, 2439–2448. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- D’alesio, C.; Bellese, G.; Gagliani, M.C.; Aiello, C.; Grasselli, E.; Marcocci, G.; Bisio, A.; Tavella, S.; Daniele, T.; Cortese, K.; et al. Cooperative antitumor activities of carnosic acid and Trastuzumab in ERBB2+ breast cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 154. [Google Scholar] [CrossRef]
- Feligioni, M.; Holman, D.; Haglerod, C.; Davanger, S.; Henley, J.M. Ultrastructural Localisation and Differential Agonist-Induced Regulation of AMPA and Kainate Receptors Present at the Presynaptic Active Zone and Postsynaptic Density. J. Neurochem. 2006, 99, 549–560. [Google Scholar] [CrossRef]
- Nisticò, R.; Florenzano, F.; Mango, D.; Ferraina, C.; Grilli, M.; Di Prisco, S.; Nobili, A.; Saccucci, S.; D’Amelio, M.; Morbin, M.; et al. Presynaptic c-Jun N-terminal Kinase 2 regulates NMDA receptor-dependent glutamate release. Sci. Rep. 2015, 5, srep09035. [Google Scholar] [CrossRef]
- Olivero, G.; Cisani, F.; Marimpietri, D.; Di Paolo, D.; Gagliani, M.C.; Podestà, M.; Cortese, K.; Pittaluga, A. The Depolarization-Evoked, Ca2+-Dependent Release of Exosomes from Mouse Cortical Nerve Endings: New Insights Into Synaptic Transmission. Front. Pharmacol. 2021, 12, 670158. [Google Scholar] [CrossRef]
- Pittaluga, A. Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory. Int. J. Mol. Sci. 2019, 20, 3641. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef]
- Langer, S.Z. Presynaptic Autoreceptors Regulating Transmitter Release. Neurochem. Int. 2008, 52, 26–30. [Google Scholar] [CrossRef]
- Olivero, G.; Bonfiglio, T.; Vergassola, M.; Usai, C.; Riozzi, B.; Battaglia, G.; Nicoletti, F.; Pittaluga, A. Immuno-pharmacological characterization of group II metabotropic glutamate receptors controlling glutamate exocytosis in mouse cortex and spinal cord. Br. J. Pharmacol. 2017, 174, 4785–4796. [Google Scholar] [CrossRef] [PubMed]
- Cisani, F.; Olivero, G.; Usai, C.; Van Camp, G.; Maccari, S.; Morley-Fletcher, S.; Pittaluga, A.M. Antibodies Against the NH2-Terminus of the GluA Subunits Affect the AMPA-Evoked Releasing Activity: The Role of Complement. Front. Immunol. 2021, 12, 586521. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.B.; Gothert, M. 5-HT receptor regulation of neurotransmitter release. Pharmacol. Rev. 2007, 59, 360–417, Erratum in Pharmacol. Rev. 2008, 60, 142. [Google Scholar] [CrossRef]
- Westphalen, R.I.; Krivitski, M.; Amarosa, A.; Guy, N.; Hemmings, H.C. Reduced inhibition of cortical glutamate and GABA release by halothane in mice lacking the K+ channel, TREK-1. Br. J. Pharmacol. 2007, 152, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, R.I.; Kwak, N.-B.; Daniels, K.; Hemmings, H.C., Jr. Regional differences in the effects of isoflurane on neurotransmitter release. Neuropharmacology 2011, 61, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, M.; Brawek, B.; Freiman, T.M.; Jackisch, R.; Feuerstein, T.J. Effects of antiepileptic drugs on glutamate release from rat and human neocortical synaptosomes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef]
- Pittaluga, A. Presynaptic release-regulating NMDA receptors in isolated nerve terminals: A narrative review. Br. J. Pharmacol. 2021, 178, 1001–1017. [Google Scholar] [CrossRef]
- Luccini, E.; Romei, C.; Raiteri, L. Glycinergic nerve endings in hippocampus and spinal cord release glycine by different mechanisms in response to identical depolarizing stimuli. J. Neurochem. 2008, 105, 2179–2189. [Google Scholar] [CrossRef]
- Stigliani, S.; Zappettini, S.; Raiteri, L.; Passalacqua, M.; Melloni, E.; Venturi, C.; Tacchetti, C.; Diaspro, A.; Usai, C.; Bonanno, G. Glia re-sealed particles freshly prepared from adult rat brain are competent for exocytotic release of glutamate. J. Neurochem. 2006, 96, 656–668. [Google Scholar] [CrossRef]
- López-Corcuera, B.; Martínez-Maza, R.; Núñez, E.; Roux, M.; Supplisson, S.; Aragón, C. Differential properties of two stably expressed brain-specific glycine transporters. J. Neurochem. 1998, 71, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.; Poyatos, I.; Aragón, C.; Gimenez, C.; Zafra, F. Characterization of the 5′ region of the rat brain glycine transporter GLYT2 gene: Identification of a novel isoform. Neurosci. Lett. 1998, 242, 25–28. [Google Scholar] [CrossRef]
- Evans, J.; Herdon, H.; Cairns, W.; O’Brien, E.; Chapman, C.; Terrett, J.; Gloger, I. Cloning, functional characterisation and population analysis of a variant form of the human glycine type 2 transporter. FEBS Lett. 1999, 463, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Armsen, W.; Himmel, B.; Betz, H.; Eulenburg, V. The C-terminal PDZ-ligand motif of the neuronal glycine transporter GlyT2 is required for efficient synaptic localization. Mol. Cell. Neurosci. 2007, 36, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Glycine and N-methyl-D-aspartate receptors: Physiological significance and possible therapeutic applications. Pharmacol. Rev. 1998, 50, 597–664. [Google Scholar]
- Nong, Y.; Huang, Y.-Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Eulenburg, V.; Hülsmann, S. Synergistic Control of Transmitter Turnover at Glycinergic Synapses by GlyT1, GlyT2, and ASC-1. Int. J. Mol. Sci. 2022, 23, 2561. [Google Scholar] [CrossRef] [PubMed]
- Yee, B.K.; Balic, E.; Singer, P.; Schwerdel, C.; Grampp, T.; Gabernet, L.; Knuesel, I.; Benke, D.; Feldon, J.; Mohler, H.; et al. Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J. Neurosci. 2006, 26, 3169–3181. [Google Scholar] [CrossRef]
- Singer, P.; Yee, B.; Feldon, J.; Iwasato, T.; Itohara, S.; Grampp, T.; Prenosil, G.; Benke, D.; Möhler, H.; Boison, D. Altered mnemonic functions and resistance to N-METHYL-d-Aspartate receptor antagonism by forebrain conditional knockout of glycine transporter 1. Neuroscience 2009, 161, 635–654. [Google Scholar] [CrossRef]
- Zafra, F.; Aragon, C.; Olivares, L.; Danbolt, N.; Gimenez, C.; Storm-Mathisen, J. Glycine transporters are differentially expressed among CNS cells. J. Neurosci. 1995, 15, 3952–3969. [Google Scholar] [CrossRef]
- Zafra, F.; Gomeza, J.; Olivares, L.; Aragón, C.; Giménez, C. Regional Distribution and Developmental Variation of the Glycine Transporters GLYT1 and GLYT2 in the Rat CNS. Eur. J. Neurosci. 1995, 7, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, I.; Ponce, J.; Aragón, C.; Giménez, C.; Zafra, F. The glycine transporter GLYT2 is a reliable marker for glycine-immunoreactive neurons. Brain Res. Mol. Brain Res. 1997, 49, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Gomeza, J.; Ohno, K.; Hülsmann, S.; Armsen, W.; Eulenburg, V.; Richter, D.W.; Laube, B.; Betz, H. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron 2003, 40, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Aragón, C.; López-Corcuera, B. Glycine transporters: Crucial roles of pharmacological interest revealed by gene deletion. Trends Pharmacol. Sci. 2005, 26, 283–286. [Google Scholar] [CrossRef]
- Eulenburg, V.; Armsen, W.; Betz, H.; Gomeza, J. Glycine transporters: Essential regulators of neurotransmission. Trends Biochem. Sci. 2005, 30, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Luque, J.M.; Nelson, N.; Richards, J.G. Cellular expression of glycine transporter 2 messenger RNA exclusively in rat hindbrain and spinal cord. Neuroscience 1995, 64, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Studler, B.; Arabadzisz, D.; Schweizer, C.; Ahmadi, S.; Layh, B.; Bösl, M.R.; Fritschy, J.M. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J. Comp. Neurol. 2004, 482, 123–141. [Google Scholar] [CrossRef]
- Danglot, L.; Rostaing, P.; Triller, A.; Bessis, A. Morphologically identified glycinergic synapses in the hippocampus. Mol. Cell. Neurosci. 2004, 27, 394–403. [Google Scholar] [CrossRef]
- Song, W.; Chattipakorn, S.C.; McMahon, L.L. Glycine-gated chloride channels depress synaptic transmission in rat hippocampus. J. Neurophysiol. 2006, 95, 2366–2379. [Google Scholar] [CrossRef]
- Romei, C.; Raiteri, L. Advances in understanding the functions of native GlyT1 and GlyT2 neuronal glycine transporters. Neurochem. Int. 2016, 99, 169–177. [Google Scholar] [CrossRef]
- Aroeira, R.I.; Vaz, S.H.; Sebastião, A.M.; Valente, C.A. BDNF modulates glycine uptake in hippocampal synaptosomes by decreasing membrane insertion of glycine transporter 2. Neurochem. Int. 2016, 99, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Aroeira, R.I.; Sebastião, A.M.; Valente, C.A. GlyT1 and GlyT2 in brain astrocytes: Expression, distribution and function. Brain Struct Funct. 2014, 219, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Aroeira, R.I.; Sebastião, A.M.; Valente, C.A. BDNF, via truncated TrkB receptor, modulates GlyT1 and GlyT2 in astrocytes. Glia 2015, 63, 2181–2197. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Armsen, W.; Papadopoulos, T.; Betz, H.; Eulenburg, V. Generation of a Mouse Line Expressing Cre Recombinase in Glycinergic Interneurons. Genesis 2010, 48, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Roux, M.J.; Supplisson, S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Supplisson, S.; Roux, M.J. Why glycine transporters have different stoichiometries. FEBS Lett. 2002, 529, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Levi, G. Cerebral amino acid transport in vitro during development: A kinetic analysis. Arch. Biochem. Biophys. 1970, 138, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Debler, E.A.; Lajtha, A. High-affinity transport of gamma-aminobutyric acid, glycine, taurine, L-aspartic acid, and L-glutamic acid in synaptosomal (P2) tissue: A kinetic and substrate specificity analysis. J Neurochem. 1987, 48, 1851–1856. [Google Scholar] [CrossRef]
- Bonanno, G.; Pittaluga, A.; Fedele, E.; Fontana, G.; Raiteri, M. Glutamic acid and gamma-aminobutyric acid modulate each other’s release through heterocarriers sited on the axon terminals of rat brain. J. Neurochem. 1993, 61, 222–230. [Google Scholar] [CrossRef]
- Kew, J.N.C.; Richards, J.G.; Mutel, V.; Kemp, J.A. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J. Neurosci. 1998, 18, 1935–1943. [Google Scholar] [CrossRef]
- Luccini, E.; Musante, V.; Neri, E.; Raiteri, M.; Pittaluga, A. N-methyl-D-aspartate autoreceptors respond to low and high agonist concentrations by facilitating, respectively, exocytosis and carrier-mediated release of glutamate in rat hippocampus. J. Neurosci. Res. 2007, 85, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, K.; Lebrun, B.; Yasuda-Kamatani, Y.; Sakaitani, M.; Shigeri, Y.; Yumoto, N.; Nakajima, T. DL-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol. Pharmacol. 1998, 53, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Barbour, B.; Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 1993, 11, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, N.X.; Granger, A.J.; Sabatini, B.L. Mechanisms and functions of GABA co-release. Nat. Rev. Neurosci. 2016, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, R. The dual glutamatergic–GABAergic phenotype of hippocampal granule cells. Trends Neurosci. 2005, 28, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, V. Co-localization of excitatory and inhibitory transmitters in the brain. Acta Neurol. Scand. 2008, 117 (Suppl. 188), 29–33. [Google Scholar] [CrossRef]
- Münster-Wandowski, A.; Zander, J.-F.; Richter, K.; Ahnert-Hilger, G. Co-existence of Functionally Different Vesicular Neurotransmitter Transporters. Front. Synaptic Neurosci. 2016, 8, 4. [Google Scholar] [CrossRef]
- Kim, S.; Sabatini, B.L. Analytical approaches to examine gamma-aminobutyric acid and glutamate vesicular co-packaging. Front. Synaptic Neurosci. 2023, 14, 1076616. [Google Scholar] [CrossRef]
- Fattorini, G.; Ciriachi, C.; Conti, F. Few, Activity-Dependent, and Ubiquitous VGLUT1/VGAT Terminals in Rat and Mouse Brain. Front. Cell. Neurosci. 2017, 11, 229. [Google Scholar] [CrossRef]
- Upmanyu, N.; Jin, J.; von der Emde, H.; Ganzella, M.; Bösche, L.; Malviya, V.N.; Zhuleku, E.; Politi, A.Z.; Ninov, M.; Silbern, I.; et al. Colocalization of different neurotransmitter transporters on synaptic vesicles is sparse except for VGLUT1 and ZnT3. Neuron 2022, 110, 1483–1497.e7. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.X.; Lima, I.V.d.A.; da Costa, F.L.P.; Rosa, D.V.; Mendes-Goulart, V.A.; Resende, R.R.; Romano-Silva, M.A.; de Oliveira, A.C.P.; Gomez, M.V.; Gomez, R.S. Glycine transporters type 1 inhibitor promotes brain preconditioning against NMDA-induced excitotoxicity. Neuropharmacology 2015, 89, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.; Artoul, S.; Segal, A.C.; Kolodney, G.; Radzishevsky, I.; Dikopoltsev, E.; Foltyn, V.N.; Inoue, R.; Mori, H.; Billard, J.-M.; et al. Neuronal d-serine and glycine release via the asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 2013, 33, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Glycine transport inhibitors in the treatment of schizophrenia. Handb. Exp. Pharmacol. 2012, 213, 367–399. [Google Scholar] [CrossRef]
- Huang, C.-C.; Wei, I.-H.; Huang, C.-L.; Chen, K.-T.; Tsai, M.-H.; Tsai, P.; Tun, R.; Huang, K.-H.; Chang, Y.-C.; Lane, H.-Y.; et al. Inhibition of glycine transporter-I as a novel mechanism for the treatment of depression. Biol. Psychiatry 2013, 74, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.J. Glycine transporter-I inhibitors: A new class of antidepressant? Biol. Psychiatry 2013, 74, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Ryan, R.M.; Carland, J.E.; Imlach, W.L.; Christie, M.J. Glycine transport inhibitors for the treatment of pain. Trends Pharmacol. Sci. 2014, 35, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-Y.; van Vliet, E.A.; Bright, K.-A.; Hanthorn, M.; Lytle, N.K.; Gorter, J.; Aronica, E.; Boison, D. Glycine transporter 1 is a target for the treatment of epilepsy. Neuropharmacology 2015, 99, 554–565. [Google Scholar] [CrossRef]
- Burgos, C.F.; Yévenes, G.E.; Aguayo, L.G. Structure and Pharmacologic Modulation of Inhibitory Glycine Receptors. Mol. Pharmacol. 2016, 90, 318–325. [Google Scholar] [CrossRef]
- Zafra, F.; Ibáñez, I.; Bartolomé-Martín, D.; Piniella, D.; Arribas-Blázquez, M.; Giménez, C. Glycine Transporters and Its Coupling with NMDA Receptors. Adv. Neurobiol. 2017, 16, 55–83. [Google Scholar] [CrossRef]
- Söderpalm, B.; Lidö, H.H.; Ericson, M. The Glycine Receptor-A Functionally Important Primary Brain Target of Ethanol. Alcohol Clin. Exp. Res. 2017, 41, 1816–1830. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, A.; Neumann, E.; Kötter, V.; Hermanns, H.; Werdehausen, R.; Eulenburg, V. The GlyT1 inhibitor bitopertin ameliorates allodynia and hyperalgesia in animal models of neuropathic and inflammatory pain. Front. Mol. Neurosci. 2018, 10, 438. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Acuña, M.A.; Gingras, J.; Yévenes, G.E. Glycine receptors and glycine transporters: Targets for novel analgesics? Cell. Mol. Life Sci. 2018, 75, 447–465. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G., Jr. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortese, K.; Gagliani, M.C.; Raiteri, L. Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals. Biomedicines 2023, 11, 3152. https://doi.org/10.3390/biomedicines11123152
Cortese K, Gagliani MC, Raiteri L. Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals. Biomedicines. 2023; 11(12):3152. https://doi.org/10.3390/biomedicines11123152
Chicago/Turabian StyleCortese, Katia, Maria Cristina Gagliani, and Luca Raiteri. 2023. "Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals" Biomedicines 11, no. 12: 3152. https://doi.org/10.3390/biomedicines11123152
APA StyleCortese, K., Gagliani, M. C., & Raiteri, L. (2023). Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals. Biomedicines, 11(12), 3152. https://doi.org/10.3390/biomedicines11123152