Abstract
The development of opioid tolerance in patients on long-term opioid analgesic treatment is an unsolved matter in clinical practice thus far. Dose escalation is required to restore analgesic efficacy, but at the price of side effects. Intensive research is ongoing to elucidate the underlying mechanisms of opioid analgesic tolerance in the hope of maintaining opioid analgesic efficacy. N-Methyl-D-aspartate receptor (NMDAR) antagonists have shown promising effects regarding opioid analgesic tolerance; however, their use is limited by side effects (memory dysfunction). Nevertheless, the GluN2B receptor remains a future target for the discovery of drugs to restore opioid efficacy. Mechanistically, the long-term activation of µ-opioid receptors (MORs) initiates receptor phosphorylation, which triggers β-arrestin-MAPKs and NOS-GC-PKG pathway activation, which ultimately ends with GluN2B receptor overactivation and glutamate release. The presence of glutamate and glycine as co-agonists is a prerequisite for GluN2B receptor activation. The extrasynaptic localization of the GluN2B receptor means it is influenced by the glycine level, which is regulated by astrocytic glycine transporter 1 (GlyT1). Enhanced astrocytic glycine release by reverse transporter mechanisms as a consequence of high glutamate levels or unconventional MOR activation on astrocytes could further activate the GluN2B receptor. GlyT1 inhibitors might inhibit this condition, thereby reducing opioid tolerance.
1. Introduction
Looking for safe analgesics and adjuncts and repurposing existing medications to treat different pain entities are still challenges in pain research. The medicinal arsenal for treating mild to severe forms of acute pain includes non-steroidal anti-inflammatory drugs (NSAIDs), minor analgesics, such as paracetamol, and opioid agonists. In this regard, the general guide known as the WHO ladder, which can assist clinicians in treating cancer pain, includes these analgesics; however, opioid types are indispensable medications in this ladder, specifically when the intensity and frequency of pain is increased and cannot be adequately controlled by NSAIDs or other minor analgesics. Indeed, there is no debate on the effectiveness of opioid analgesics in relation to the management of cancer pain or noncancer chronic pain at the initiation of therapy. In the short-term treatment of pain, adverse effects related to the gastrointestinal tract develop, but generally, they are treatable with antiemetics, laxatives, and other drugs [1]). However, with long-term opioid analgesic treatment, the development of analgesic tolerance creates an obstacle, and to overcome it, dose escalation is required. Undoubtedly, further dose escalation results in benefits and risks for patients. Opioid tolerance occurs after long and repeated treatment with opioid agonists and is defined as a decrease in opioid potency, which is reflected by an increase in ED50 or EC50 values. These values are indicative of a pharmacological shift to the right in the dose/concentration–response curves of test opioid agonists, such as morphine [2,3]. In noncancer chronic pain, opioid analgesic tolerance does also occur, and the consequence of opioid dose escalation is that the patient is exposed to the risk of adverse events, including overdose and opioid use disorders (OUD) as well as other opioid side effects, as mentioned above [4,5,6]. Many preclinical and clinical studies have been conducted to understand the underlying mechanisms of opioid analgesic tolerance. In fact, µ-opioid receptors (MORs) mediate the analgesic effect of the most used opioid agonists in clinical practice. These indices indicate that opioid analgesic tolerance mediated through MORs is a clinical drawback hindering long-term opioid treatment. Furthermore, pharmacological blockade of spinal MORs with opioid antagonists, such as naloxone or H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) (a highly selective MOR antagonist), abolishes the analgesic effect of systemic morphine and fentanyl [7]. This means that elucidating spinal mechanisms that contribute to opioid analgesic tolerance could provide new knowledge that would lead to developing novel drug classes or at least adjuvants to taper off opioid doses. Indeed, there are many proposed mechanisms of opioid analgesic tolerance development, but none of them have brought significant value in the clinical setting. However, the diverse mechanisms related to opioid analgesic tolerance offer future research avenues that might ultimately unlock the solution to opioid tolerance. At the signal transduction level, MORs cause desensitization and downregulation (see Table 1), yet compensatory/opponent processes, such as neuroadaptations, occurred upon long-term exposure to opioid analgesics, such as morphine [3,8]. These processes encompass consequences related to opioid analgesic efficacy and homeostatic adaptation changes (opposite effect e.g., hyperalgesia). In this regard, the dose escalation effect is not only limited to the above-mentioned side effects, including opioid analgesic tolerance, but also can precipitate or exacerbate opioid-induced hyperalgesia [3]. The most promising achievements related to counteracting opioid analgesic tolerance was the application of competitive and non-competitive antagonists of ionotropic N-methyl-D-aspartate acid glutamate receptors (NMDARs), namely, MK-801 (dizocilpine), ketamine, and phencyclidine (PCP). In this regard, a large amount of evidence on the contribution of these receptors to opioid analgesic tolerance has been published in recent decades [9]. Alongside these studies, additional papers have also focused on the impact of spinal glutamate receptors in opioid analgesic tolerance, particularly with NMDARs, which are located on the pre- and postsynaptic membrane [10]. Furthermore, there are studies describing compounds that inhibit opioid analgesic tolerance by blocking GluN2A-2D receptor-operated ion channels, such as MK-801 [11,12]. However, these inhibitors bear several side effects, including cognitive impairment and dissociative behaviors [13,14]. Drugs devoid of these side effects that directly or indirectly affect the function of GluN2A-2D-receptor-operated ion channels in relation to opioid analgesic tolerance would be worth investigating in the future.

Table 1.
The main enzymes, proteins, and mechanisms involved in opioid analgesic tolerance.
In this review, we shed light on undiscovered targets in opioid analgesic tolerance, namely glycine transporter type 1 (GlyT1). The prediction is supported by the following evidence from the literature: (i) GlyT1 regulates extrasynaptic glycine concentrations; (ii) glycine acts as a co-agonist at GluN2A-2DRs; (iii) GluN2B is predominantly found in the extrasynaptic region; and (iv) GluN2BR plays a crucial role in the development of opioid analgesic tolerance.
2. N-Methyl-D-aspartate Acid Glutamate Receptors as Key Players in the Development of Opioid Analgesic Tolerance
As mentioned in the introduction, in recent decades, extensive research has been conducted to elucidate the underlying mechanisms of opioid analgesics tolerance at different pharmacodynamic levels. At the cellular level, several postulated mechanisms have been proposed for opioid analgesic tolerance, as described in Table 1, though the exact mechanisms responsible are poorly understood. With respect to receptor desensitization and opioid analgesic tolerance development, several mechanisms have been proposed, including one that relies on the activation of NMDARs [23,24,25]. This mechanism is supported by the fact that MK-801, a highly potent and selective noncompetitive NMDAR antagonist, attenuates the development of morphine analgesic tolerance in mice [12,26]. Further preclinical studies have shown that opioid tolerance that developed upon long-term treatment with intrathecal morphine is attenuated by MK-801 or 5-AP, competitive NMDAR antagonist, or GM1 ganglioside (an intracellular PKC inhibitor) in rats [27,28]. All these data shed light on the involvement of glutamate receptors in the development of opioid analgesic tolerance. The next section focuses on deciphering the receptors that mediate the glutamate effects related to opioid analgesic tolerance, particularly in the spinal cord.
2.1. Metabotropic Glutamate and NMDA-Type Glutamate Receptors
Glutamate receptors are classified as metabotropic glutamate receptors (mGluRs) [29,30] (Table 2) and ionotropic glutamate receptors (iGluRs) [31]. They belong to the G-protein coupled receptor and tetrameric ionotropic receptor superfamilies, respectively. Based on the activator ligands, iGluRs are further classified as NMDA (Table 3), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid), and kainate receptors [32]. Owing to the signaling pathway feature, metabotropic receptors mediate slower responses when compared to ionotropic receptors [33]. The mGluRs include eight subtypes, named mGluR1 through mGluR8 [34]. Furthermore, based on the homology of amino acid sequences, agonist selectivity, and their interactions with intracellular second messengers, mGluRs are further classified into three groups, namely groups I, II, and III. The group I receptors belong to G protein-coupled receptors, namely Gaq/11, and mediate their effects through inositol phosphate 3/Ca2+ (PLC) signal transduction. Likewise, groups II and III also belong to G protein-coupled receptors, but they couple to Gi/o type G-proteins, and the signal transduction mechanism relies on the inhibition of adenylate cyclase. Almost all mGluRs, except mGluR6, show a ubiquitous expression pattern in the CNS both in neurons and glial cells, though some subtypes have specific regional expression, as described in Table 2. The group I (mGluR1 and mGluR5), group II (mGluR2 and mGluR3), and group III (mGlu4,6,7, and 8) receptors are activated by 3,5-dihydroxyphenylglycin (3,5-DHPG), LY354740, and L-2-amino-4-phosphonobutyrate (L-AP4), respectively [34,35]. The group II receptors (mGluR3-LI) have been reported to show a pattern of distribution in the brain and spinal cord areas related to pain processing. In this regard, in the brain stem and spinal cord, the distribution was weak, whereas in the principle sensory trigeminal nucleus, spinal trigeminal nuclei, paratrigeminal nucleus, and spinal cord dorsal horn, the distribution was moderate or strong [35,36]. With respect to pain control related to group I mGluRs, several preclinical studies have shown that the activation of these receptors results in pronociceptive action in different animal pain models [37,38,39,40]. Since the objective of the present work was to map the contributions of GlyT1 to the development of opioid analgesic tolerance, extrasynaptically localized NMDARs are reviewed in the next section.
Table 2.
Distribution of metabotropic glutamate receptors in central nervous system areas related to pain processing.
2.2. Glutamatergic Ionotropic NMDA Receptors and Opioid Analgesic Tolerance
NMDARs belong to the ligand-gated ion channel family and mediate glutamate neurochemical transmission, as described in the previous section. These receptors have a tetrameric structure, and each subtype is composed of different subunits, including GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A, and GluN3B, as shown in Table 3. NMDARs’ macromolecular structures are built up from four subunits designated as GluN1 and GluN2A-D. These heterotetrameric structures host two copies of GluN1 (obligatory) and two copies of one of the GluN2s (GluN2A, 2B, 2C, or 2D) [31]. Thus, the NMDA-operated ion channels exhibit different characteristics that result in functional diversity [53]. The two GluN2 subunits (an orthosteric binding site) bind glutamate, and the two GluN1 subunits (co-agonist binding sites) bind glycine or D-serine (Table 4). An example for such diversity is a tetramer of 2GluN1-2GluN2B (heterotetramer).
The opening of the NMDA receptor-operated channel is unique and complex. It requires the simultaneous occupation of the agonist binding site by glutamate and the co-agonist binding sites either by glycine or D-serine, along with the depolarization of cells to remove Mg2+ blockade. NMDA-operated ion channels are non-ion selective; in addition to Ca2+, they allow for the entry of monovalent cations, such as Na+ and K+, into the cells. It has been proposed that the co-agonist binding site of NMDAR is equivalent to the second agonist binding site but was altered during evolution. The pattern distribution and the subclasses of glutamatergic ionotropic NMDARs are presented in Table 3. Several data suggest that GluN2B receptors are predominantly localized extrasynaptically; see Table 3 and Traynelis et al. [54].
Overactive GluN2B receptors are thought to play a key role in analgesic tolerance elicited by the repeated administration of opioid agonists [55]. On the contrary, different pharmacological interventions, which decrease NMDAR overactivity, inhibit the development of opioid tolerance in analgesia. Shimoyama and coworkers [56] reported that the NMDAR channel blockers ketamine and MK-801 can suspend the analgesic tolerance of opioids. These data are in line with that above mentioned (see Section 2) and reviewed in the literature [57,58,59,60,61,62]. It has also been reported that the negative allosteric modulators of the GluN2B receptor, such as ifenprodil or Ro25-6981, reduced NMDAR activity and suspended the development of opioid tolerance in nociception [63].
Table 3.
Distribution of N-methyl-D-aspartate acid glutamate receptor family in central nervous system areas related to pain processing.
Table 3.
Distribution of N-methyl-D-aspartate acid glutamate receptor family in central nervous system areas related to pain processing.
| Glutamate Receptor Subunits | Area (Region) | Distribution Pattern | Subject | References |
|---|---|---|---|---|
| GluN1 (NMDAR1) | GluN1-2: widely distributed in the CNS GluN1-1: in rostral regions (e.g., cortex) GluN1-4: in caudal regions (e.g., brainstem) Dorsal horn of spinal cord Spinal cord Spinal cord: laminae I–III | +++ +++ +++ +++ +++ +++ | Rat | [64,65,66,67,68] |
| GluN2A (NMDAR2A) | Cerebral cortex Substantia gelatinosa neurons (synaptic localization) Spinal cord | +++ +++ | Rat | [67,69,70] |
| GluN2B (NMDAR2B) | Telencephalon and thalamus DRG neurons (primary afferent neurons) Substantia gelatinosa neurons (extrasynaptic localization) Spinal cord: laminae I–III Spinal cord: lamina II (and IX) | +++ +++ * ++/+++ +/++ | Rat | [67,68,69,70,71] |
| GluN2C (NMDAR2C) | All regions except cerebellar cortex Spinal cord: laminae I–III Spinal cord | + 0 0 | Rat | [67,68,69] |
| GluN2D (NMDAR2D) | Brainstem, cortex Substantia gelatinosa neurons Spinal cord | + * 0 | Rat | [67,69,70] |
| GluN3A (NR3A) | Thalamus (VPL) Cervical spinal cord: in proximity to the dorsal horn | ++ +++ | Rat (postnatal day 16) | [72] |
| GluN3B (NR3B) | Cerebral cortex Spinal cord: laminae I–II (and VIII-IX) (**) Spinal cord: laminae III–VI | +++ ++/+++ | Rat | [73] |
+ The degree of distribution pattern; * electrophysiological study; ** in the dorsal horn, parvalbumin-positive interneurons are NR3B-positive (these neurons are glycinergic interneurons). IUPHAR terminology used, with previously used nomenclature in brackets.
Table 4.
Endogenous and exogenous modulators for N-methyl-D-aspartate acid glutamate receptors [31].
Table 4.
Endogenous and exogenous modulators for N-methyl-D-aspartate acid glutamate receptors [31].
| Glutamate Re-ceptor Subunits | Endogenous Agonists | Exogenous Agonists | Exogenous Antagonists | Channel Blocker | |||
|---|---|---|---|---|---|---|---|
| Glutamate Site | Glycine Site | Glutamate Site | Glycine Site | Glutamate Site | Glycine Site | ||
| GluN1 (NMDAR1) | L and D-Asp | Glycine D-serine | NMDA HQA * | (+)-HA966 * | - | 5,7-DCKA | |
| GluN2A (NMDAR2A) | L and D-Asp | Glycine D-serine | NMDA HQA * | (+)-HA966 * | D-AP5 | 5,7-DCKA | Mg2+, MK-801, ketamine, phencyclidine, amantadine. |
| GluN2B (NMDAR2B) | L and D-Asp | Glycine D-serine | NMDA HQA * | (+)-HA966 * | D-AP5 | 5,7-DCKA | Mg2+, MK-801, ketamine, Phencyclidine, amantadine. |
| GluN2C (NMDAR2C) | L and D-Asp | Glycine D-serine | NMDA HQA * | - | D-AP5 | 5,7-DCKA | Phencyclidine, ketamine, amantadine, Mg2+, MK-801. |
| GluN2D (NMDAR2D) | L and D-Asp | Glycine D-serine | NMDA HQA * | - | D-AP5 | 5,7-DCKA | Mg2+, MK-801, amantadine, ketamine, phencyclidine. |
| GluN3A (NR3A) | |||||||
| GluN3B (NR3B) | |||||||
* Electrophysiological study; aspartic acid (Asp); homoquinolinic acid (HQA); 5,7-Dichlorokynurenic acid (DCKA).
3. Glycine Transporter Type 1 and N-Methyl-D-aspartate Acid Glutamate Receptors: Their Locations and Functions in the Glial–Neural Tripartite Synapse
We have hypothesized interactions among GlyT1, NMDA, and opioid receptors in the development of opioid analgesic tolerance. The morphological basis for this interaction is the tripartite model, which consists of an astrocytic process, a presynaptic axon terminal, and a postsynaptic element with the synaptic cleft [74]. It has been demonstrated that MORs are present on glutamatergic axon terminals and astrocytes [75]. NMDARs are expressed in both pre- and postsynaptic neurons, and GlyT1 is localized in astrocytes and in glutamatergic axon terminals interacting with NMDA-type glutamate receptors [76]. However, these receptors and transporters are present in the glial–neural tripartite synapse in various forms.
Typical and atypical MORs are coupled either to the conventional Gi signal transduc-tion or, in the unconventional form, to Gq protein. These receptors are operated with cAMP and PKA phosphorylation or DAG/IP3 and PKC-mediated phosphorylation. In controlling analgesia, these MORs operate in glutamatergic axon terminals or astroglia cells, respectively [75]
GluN2B receptors are predominantly localized extrasynaptically. This localization creates possibilities for glutamates diffused out from neighboring synaptic clefts or astrocytes to influence the function of GluN2B receptors [77]. This pattern of neuronal organization occurs in the spinal cord, cortex, and thalamus [78]. On the other hand, GluN2A receptors have been suggested to be synaptically expressed. The prerequisite for the activation of the formerly mentioned receptors is the simultaneous presence of the following three components: glutamate, co-agonists (either glycine or D-serine), and the removal of Mg2+ from NMDA receptor ion channels by depolarization.
Thirdly, GlyT1 can primarily be detected with glial fibrillary acidic protein (GFAP) in astroglia cells and has a role in building up extrasynaptic glycine concentrations by bi-directional operation [79,80]. GlyT1 consists of 12 transmembrane (TM) domains. TM1-5 and TM6-10 form a gate for 2Na+ and 1Cl− ion transport [81]. In addition to glial GlyT1, this transporter was also reported to be present in the vicinity of GluN2B receptors (GluN2BRs) in the membranes of postsynaptic neurons. Neural GlyT1 was suggested to assure the co-agonist glycine for this receptor activation [82]. To understand the mechanisms and the function of spinal glycine homeostasis, the next section sheds light on ligands affecting GlyTs.
3.1. Classification of Glycine Transporter Inhibitors
As we attempt to build a hypothesis on how the tripartite model is involved in the development of opioid analgesic tolerance, it is essential to review spinal glycine homeostasis, particularly in the vicinity of GluN2BRs. This means that GluN2BRs, GlyTs, and MORs would contribute to the development of opioid analgesic tolerance. Nevertheless, GlyT1 would be of interest because it plays a fundamental role in the regulation of extrasynaptic glycine levels [24,83,84]. In this regard, we summarize the various GlyT1 and GlyT2 inhibitors to help the readers understand the entire spinal glycine regulatory system in the hope of making successful predictions about the development of opioid analgesic tolerance.
Several GlyT inhibitors have been described in the last two decades. Although they selectively inhibit either astrocytic GlyT1 or neuronal GlyT2, animal models of human disorders have also pointed to the need for non-selective GlyT inhibitors [85]. Modeling neuropathic pain in rats has indicated the involvement of both types of GlyTs in the pathological alterations of spinal cord neuronal circuitries [85,86,87,88].
At present, GlyT inhibitors can be classified based on their chemical structures. The very first GlyT1 inhibitor that was discovered was glycyldodecylamide (GDA), which exhibited modest inhibitory potency [89]. This compound called attention to the importance of membrane lipids around GlyT1 in the binding of ligands to the transporter molecule. The amide-head in GDA suggests a possible ionic bonding interaction of this compound with Tyr128 in the transporter [90]. Compounds derived from the endogenous glycine uptake inhibitor arachidonic acid also indicate the importance of the lipid tail, which may interact with the lipid environment of the cell membrane. In addition to the lipophilic part, oleyl-L-carnitine also contains a basic head group [83]. Despite some similarities in the chemical structures of GDA and oleyl-L-carnitine, the former acts as an inhibitor of GlyT1, and the latter has been shown to more likely block GlyT2 activity.
While the role of the basic head group in the binding of inhibitors to GlyTs has been shown, other GlyT inhibitors contain free carboxylic groups derived from glycine or sarcosine. Sarcosine is a modest but selective GlyT1 inhibitor, with a possible binding site in the cavities formed by the Tyr128-Tyr302-Ser303-Leu304-Gly305 amino acids within the transporter molecule [90]. Although sarcosine is considered to be a competitive GlyT1 antagonist [91], we found it to exhibit a substrate-type inhibition of the transporter, shifting its operation to the reverse mode [90]. Lipid-containing GlyT2 inhibitors with a free carboxylic group instead of a basic head have also been reported (N-arachidonyl-glycine, N-oleyl-glycine) [92].
Based on the selective GlyT1 inhibitory property of sarcosine, a great number of GlyT1 inhibitors were synthesized by attaching extended lipophilic aryl groups to the N atom [93,94]. These compounds are called sarcosine-based GlyT1 inhibitors, which exhibit a competitive or non-competitive type of transporter inhibition [79,95]. Examples of these inhibitors are NFPS, Org-24461, SSR-130800, LY-2365109, and SzV-1997. Compounds from Merck and Allelix (ALX1393) containing glycine instead of sarcosine have also been synthesized and possess inhibitory effects on GlyT1 and GlyT2, respectively [96].
Because of the adverse effects of sarcosine-based GlyT1 inhibitors (often due to their pharmacokinetic properties), chemical synthesis has turned toward non-sarcosine-based GlyT1 inhibitors. A great number of compounds have also been synthesized in this series, and some of them have undergone clinical investigations as well. Of these compounds, we found the benzamide derivatives particularly interesting. Compounds containing the benzamide skeleton exhibited selective GlyT1 (ACPPB, SSR-504734, GSK-1018921) or GlyT2 (Org-25543) inhibitory properties (Figure 1). Therefore, we believe that consideration of the chemical structures of ACPPB and Org-25543 may be the basis for identifying non-selective GlyT inhibitors with a novel pharmacological profile in different experimental conditions or even in clinical use. Nevertheless, the development of selective GlyT1 inhibitors seems to be ideal for therapeutic purposes in the context of the present review, namely for opioid analgesic tolerance.
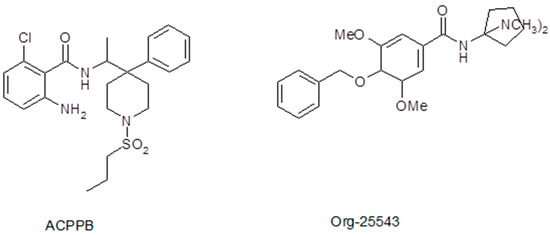
Figure 1.
Chemical structures of benzamide derivatives GlyT1 inhibitor ACPPB and GlyT2 inhibitor Org-25543. We speculate that these two compounds may be sources for synthesizing a novel non-selective GlyT1/GlyT2 inhibitor.
3.2. Compounds Acting as Ligands for Glycine Transporter Type 1 and NMDA Receptor Interactions: Some Operational Characteristics
Glycine acts as an agonist on glycine receptors (strychnine-sensitive binding site) and as a co-agonist on the strychnine-insensitive glycine binding site of GluN2A-D type NMDARs (Table 4) in the spinal cord. Glycine is also the substrate for GlyTs, and it is released from astrocytes or glycinergic nerve endings in the CNS.
Other compounds may also bind as co-agonists to the glycine binding sites of NMDARs or act as substrates for GlyT1 (Figure 2). The structural similarities between glycine and sarcosine raise the possibility that sarcosine has a co-agonist role in NMDARs [91]. As discussed above, sarcosine was one of the very first GlyT1 inhibitors to be found. This inhibition of GlyT1 may be the substrate type, eliciting [3H]glycine efflux from glia cells by shifting GlyT1 operation into the reverse mode [90]. Moreover, this effect of sarcosine, which is identical to that of the natural substrate glycine, was suspended by the addition of the GlyT1 inhibitor NFPS (Figure 3, legend).
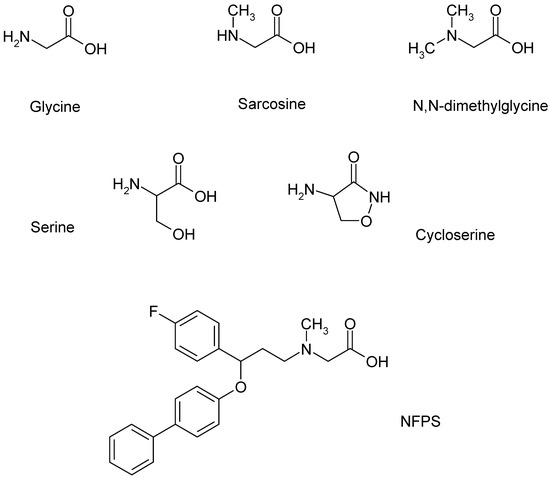
Figure 2.
Ligands of GlyT1 and NMDA receptors, which form a functional interaction in the glia cell, presynaptic axon terminal, and postsynaptic neuron tripartite model. NFPS is a sarcosine-based GlyT1 inhibitor.
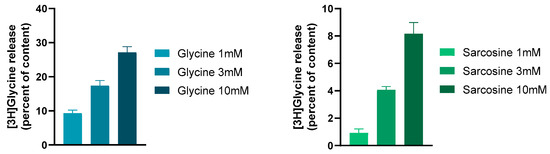
Figure 3.
The GlyT1 ligands glycine and sarcosine evoked a concentration-dependent and external Ca2+-independent release of [3H]glycine from rat hippocampal slices. This release was due to the reverse-mode operation of GlyT1. The slices were incubated with [3H]glycine, and then superfused with aerated (95% O2/5% CO2) and preheated (37 °C) Krebs bicarbonate buffer. Twenty-two 3 min fractions were collected. Drugs were added to the hippocampal slices from fraction 5 and maintained throughout the experiments. At the end of superfusion, the tissue content of radioactivity and the radioactivity released from the tissues were determined. The GlyT1 non-competitive antagonist NFPS (0.1 mM) reversed the effect of 3 mM sarcosine on [3H]glycine release: it was 0.68 ± 0.21 and 4.07 ± 0.24 per cent of the content released in the presence and absence of NFPS, respectively. Student’s t statistics for two-means, p < 0.001, mean ± S.E.M., n = 4–8. Data were taken and redrawn with permission [90].
Sarcosine, in addition to its action on GlyT1, also acts directly on NMDARs as a co-agonist [91]. Using a whole cell patch clamp recording in rat prefrontal cortex slices, we found that sarcosine exhibited a bidirectional effect. At lower concentrations, it inhibited the NMDA receptor-mediated currents, whereas, at higher concentrations, it enhanced them (see Figure 4). Moreover, these experiments also suggested a possible partial agonist effect of sarcosine on NMDA receptor-mediated currents. These findings are contradictory to those of McBain et al. [97], who reported that sarcosine did not bind to the glycine-binding site of NMDARs expressed in Xenopus oocytes. Nevertheless, Lee and colleagues [98] showed that sarcosine, in addition to its action as a GlyT1 inhibitor, also potentiated NMDAR function by acting as a co-agonist at the glycine binding site of NMDARs. Sarcosine alone does not initiate a response but enhances NMDA receptor-mediated currents when applied with glutamate [98]. What is interesting in our results is that at lower concentrations (1 and 10 μM), sarcosine reduced NMDA receptor-mediated currents (Figure 4). At present, we can explain the observed effect as follows: (i) The measured NMDA receptor-mediated currents are the result of the binding of both glutamate and glycine (present in tissue); (ii) at a low concentration, sarcosine behaves as a partial agonist on glycine binding sites, and consequently, hinders the physiological effect of glycine; (iii) at high concentrations, sarcosine inhibits GlyT1, and as a result, more glycine is available in the vicinity of NMDARs. On the other hand, it is clear that glycine is a full agonist and shows an effect on one direction (Figure 4). However, the presented data for glycine reveal a decreased effect of glycine at higher concentrations. This feature might be related to the ability of glycine to evoke NMDAR desensitization. This finding might correlate with that of Zhang and coworkers [91], who concluded that NMDAR desensitization is more likely to occur with glycine than with sarcosine.
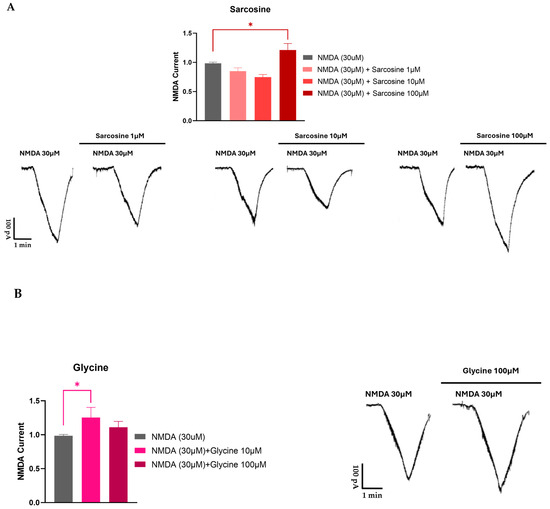
Figure 4.
Electrophysiological recording from rat prefrontal cortex slices for sarcosine (A) and glycine (B). Sarcosine induced a biphasic effect on NMDA receptor-mediated currents. Ten-day-old Wistar rat pups were decapitated, and coronal slices containing the medial prefrontal cortex (PFC) were prepared from the brain using a vibrotome. The slices were stored in a holding chamber before use. A single slice was transferred into a recording chamber and superfused (2.5–3 mL/min) with aerated artificial cerebrospinal fluid at room temperature. Whole cell patch clamp recordings were implemented. Pyramidal cells in the PFC were visualized with an upright microscope. Borosilicate glass patch pipettes were filled with standard intracellular solution; the tip resistance was 5–7 MΩ. The membrane currents were recorded in the voltage–clamp configuration of the amplifier at a holding potential of −70 mV. The data were filtered at 2 kHz with a lowpass filter of the amplifier, digitized at 5 kHz, and stored in a computer. NMDA (30 μM) was applied three times (T1, T2, and T3) for 1.5 min with 10 min intervals. Test drugs were added to the bath 5 min before and during the third application of NMDA. Since the amplitudes showed great variabilities among the cells, the effects at T3 were presented as the T3/T2 ratio. The T3/T2 ratios were summarized as the mean ± SEM of n. Statistical significance was established by one-way ANOVA, followed by Dunnett’s post hoc test. p < 0.05 was considered to be statistically significant (*).
The sarcosine analogue N,N-dimethylglycine, which did not affect GlyT1 operation in our [3H]glycine efflux experiments, also binds to the NMDA receptor glycine binding site (Table 5). The potencies of sarcosine and N,N-dimethylglycine in the brain slices were found to be equal. N,N-Dimethylglycine acts more as a partial agonist at the glycine binding site of NMDA receptors [98].
Table 5.
Ligands for glycine transporter type 1 and N-methyl-D-aspartate acid glutamate receptors.
Another glycine analog, N-ethylglycine, reduced GlyT1-dependent glycine uptake, acting as an alternative substrate for GlyT1. It is a selective and competitive inhibitor of GlyT1 without altering GlyT2 activity [99].
D-Serine acts as a co-agonist on synaptic NMDA receptors, with a subunit composition of GluN1/GluN2A receptors [100]. D-Cycloserine was reported to be a partial agonist at the glycine binding site of NMDA receptors [101].
4. Interplay between Glycine Transporter Type 1 and N-Methyl-D-aspartate Acid Glutamate Receptors in Reversal of Opioid Tolerance: A Hypothesis
To the best of our knowledge, the interaction between opioid and glycine systems in relation to opioid analgesic tolerance has not been elucidated thus far. Sufficient results are available for MOR-mediated analgesia and possible mechanisms contributing to the development of opioid analgesic tolerance, as mentioned in the introduction. With respect to analgesia, morphine reduces pain reactions by the activation of conventional MORs in the dorsal horn of the spinal cord [7], and the inhibition of these receptors by intrathecal MOR antagonists has been reported. Also, opioid agonists decrease glutamate release from glutamatergic nerve endings, and this effect may be at least part of the evoked analgesia [102]. MORs’ conventional signal transduction following opioid treatment and the consequent development of opioid tolerance encompasses mitogen-activated protein kinase (MAPKs: ERK1/2, p30, JNK) activation through β-arrestin. These kinases activate presynaptic GluN2B receptor [23]; see Figure 5.
However, the administration of morphine also activates unconventional opioid receptors expressed in astroglia cells [75,103]. These receptors are coupled to Gq proteins, and their signaling evokes an increase in the protein kinase C (PKC)-mediated phosphorylation of GlyT1. It has been shown that phorbol 12-myristate 13-acetate, a PKC activator, evokes the inhibition of [3H]glycine uptake in HEK 293 and C6 glioma cells [104,105]. The inhibition of glycine uptake by phosphorylated GlyT1 may be a similar mechanism that has been reported for other plasma membrane neurotransmitter transporters [106]. Along this line, we speculate that the PKC-mediated phosphorylation of GlyT1 inhibits the uptake-mode operation, which may be associated with an increased rate of the reverse-mode operation of the transporter [107]. The shift in the bidirectional operation of GlyT1 to the release mode evoked by PKC-mediated phosphorylation may lead to a marked increase in extracellular glycine concentrations, similar to a number of other experimental conditions [108]. An increase in extracellular glycine concentrations results in a co-agonist-induced overactivation of extracellular GluN2B receptors and strengthens the inhibition of opioid receptors, which then causes the development of opioid tolerance in analgesia. This negative influence exerted by NMDA receptors on opioid receptors occurs at the signal transduction pathways of the two receptors (NOS-guanylyl cyclase-PKG signaling) [109].
We have found that GlyT1 inhibitors block transporter operation in both directions in several experimental conditions [108]. The inhibition of the reverse-mode operation of GlyT1 leads to a decrease in extracellular glycine concentrations, reducing the co-agonist activation of the extracellular GluN2B receptor, which then results in the suspension of the negative influence on MOR activity. In a condition like this, the sensitivity of MORs to morphine or other opioid agonists may be restored, and opioid tolerance development may be delayed (Figure 5).
To further strengthen our hypothesis about a concomitant activation of MOR and the inhibition of GlyT1 as a mechanism responsible for delaying the development of opioid analgesic tolerance, in vivo studies are required to support this hypothesis.
Another possible mechanism is that long-term treatment with opioid analgesic agonists, such as morphine, causes an increase in glutamate levels in the spinal cord, as described previously [110]. An increase in glutamate levels can trigger glycine release, which in turn, together with increased glutamate, can enhance GluN2B receptor activity. This condition can give rise to the development of opioid analgesic tolerance (Figure 5A–D). The excitatory amino acid transporter (EAAT)-mediated release of D-aspartate, a drug that mimics glutamate, was lowered in the presence of GlyT1 inhibitors in synaptosomes of the spinal cord and hippocampus [111,112]. The result may be a decrease in GluN2B receptor activation, which has been described to largely be involved in opioid analgesic tolerance [113].



Figure 5.
(A,B) Chronic treatment with opioid analgesics, such as morphine, initiates MOR-mediated MAPK activation through the phosphorylation process that occurs in the presynaptic nerve ending. This activation encompasses the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun amino-terminal kinases, and p38. As a result, the GluN2A and GluN2B receptors are activated. NMDAR activation in this condition enhances glutamate release. The activation of GluN2A and GluN2B receptors requires glutamate and co-agonist (glycine). The source of glutamate is the activated neuron, whereas the source of glycine is either glycinergic neurons or astrocytes [16,23]. The illustrations were created by BioRender, with agreement numbers 2023 IS268LVHRP for 5/A and 2023 JT268LWY14 for 5/B. (C,D) A glial–neuronal tripartite model of opioid tolerance and its potential reversal by GlyT1 inhibitors. Gq protein-coupled unconventional MORs are activated by acute or repeated morphine administration in astroglia cells. The increase in IP3/DAG production and [Ca2+]i triggers PKC-mediated phosphorylation, which shifts the balance of the uptake-release mode of GlyT1 toward release-mode operation. The consequent high extracellular glycine levels upregulate extracellular NMDA GluN2B receptors, which inhibit the signal transduction of µ opioid receptors resulting in the development of opioid tolerance. GlyT1 inhibitors inhibit the bidirectional operation of the transporter and decrease elevated extracellular glycine levels, reversing NMDA receptors’ sensitivity. This adapts opioid receptor sensitivity to normal levels. Abbreviations: NOS, nitric oxide synthases; GC, guanylyl cyclase; PKG, protein kinase G. The illustrations were created by BioRender, agreement numbers 2023 IU268LVTVC for 5/C and 2023 EO268LVZVX for 5/D. Figure 5 was adapted from previous works [75,109,114].
Figure 5.
(A,B) Chronic treatment with opioid analgesics, such as morphine, initiates MOR-mediated MAPK activation through the phosphorylation process that occurs in the presynaptic nerve ending. This activation encompasses the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun amino-terminal kinases, and p38. As a result, the GluN2A and GluN2B receptors are activated. NMDAR activation in this condition enhances glutamate release. The activation of GluN2A and GluN2B receptors requires glutamate and co-agonist (glycine). The source of glutamate is the activated neuron, whereas the source of glycine is either glycinergic neurons or astrocytes [16,23]. The illustrations were created by BioRender, with agreement numbers 2023 IS268LVHRP for 5/A and 2023 JT268LWY14 for 5/B. (C,D) A glial–neuronal tripartite model of opioid tolerance and its potential reversal by GlyT1 inhibitors. Gq protein-coupled unconventional MORs are activated by acute or repeated morphine administration in astroglia cells. The increase in IP3/DAG production and [Ca2+]i triggers PKC-mediated phosphorylation, which shifts the balance of the uptake-release mode of GlyT1 toward release-mode operation. The consequent high extracellular glycine levels upregulate extracellular NMDA GluN2B receptors, which inhibit the signal transduction of µ opioid receptors resulting in the development of opioid tolerance. GlyT1 inhibitors inhibit the bidirectional operation of the transporter and decrease elevated extracellular glycine levels, reversing NMDA receptors’ sensitivity. This adapts opioid receptor sensitivity to normal levels. Abbreviations: NOS, nitric oxide synthases; GC, guanylyl cyclase; PKG, protein kinase G. The illustrations were created by BioRender, agreement numbers 2023 IU268LVTVC for 5/C and 2023 EO268LVZVX for 5/D. Figure 5 was adapted from previous works [75,109,114].



Our hypotheses focus primarily on GlyT1 operation in the development of opioid analgesic tolerance. It is also important to determine which brain areas are primarily favored for the interactions among GlyT1, NMDA-, and opioid receptors. It would also be interesting to see whether a hypothesis on the regulatory role of GlyT1 operation in opioid tolerance is valid only in analgesic tolerance or tolerance for other opioid effects also. Further elucidation of these questions is currently the prime interest of our laboratory.
5. Discussion and Conclusions
Overactive GluN2B receptors are thought to play a key role in analgesic tolerance elicited by the repeated administration of opioid analgesics. In fact, different pharmacological interventions, which decrease NMDAR overactivity, inhibit the development of opioid tolerance in analgesia. Thus, the NMDAR channel blockers ketamine and MK-801 and the negative allosteric modulators of the GluN2B receptor (ifenprodil, Ro25-6981) reduce NMDAR activity and suspend the development of opioid tolerance in nociception. Along this line, here, we suggest a potential interaction between GlyT1 and GluN2B receptors and conventional and unconventional MORs in the development of opioid analgesic tolerance. The morphological basis for these interactions is the presynaptic axon terminal, postsynaptic element with the synaptic cleft, and the astrocytic processes. These contributors, namely GlyT1 and NMDARs, are present in different forms in this tripartite model. Indeed, the functional interaction between GlyT1 and NMDARs is now generally accepted [115]. This interaction is based on the fact that extrasynaptic glycine concentrations, which are regulated by GlyT1, determine NMDAR activity by modulating their co-agonist sites. GlyT1 and NMDAR interaction may be either stimulatory or inhibitory in different CNS pathologies. In this regard, in schizophrenia, NMDARs are believed to be hypoactive; the NMDAR channel blockers PCP and ketamine worsen patients’ conditions, and GlyT1 inhibitors were developed in the hope of restoring the NMDAR hypofunction observed in this disorder [116,117,118,119,120,121]. On the contrary, NMDAR hyperfunction has been reported in depression, hypoxic/anoxic conditions, and convulsions [119,122,123,124,125]. The NMDAR channel blocker ketamine suspends NMDAR overactivity in these conditions [126,127,128], and GlyT1 inhibition exerts antidepressant, neuroprotective, and anticonvulsive effects [122,129,130]. In addition, ketamine and dizocilpine suspended opioid analgesic tolerance, suggesting NMDAR hyperfunctionality in repeated opioid receptor stimulation [56]. Owing to these literature data, we hypothesize that GlyT1 inhibitors could delay opioid analgesic tolerance development. Thus, depending on pathological conditions, NMDAR channel inhibitors and GlyT1 inhibitors may influence NMDAR activity in the identical or opposite directions. Since GlyT1 regulates the extracellular glycine concentration, reduced NMDAR activity in schizophrenia may be the result of overactive GlyT1 operation in the uptake mode. On the other hand, enforced release-mode operation of GlyT1 leads to elevated extracellular glycine concentrations causing NMDAR hyperactivity in depression, neurodegeneration, seizures, or opioid analgesic tolerance. Thus, GlyT1 inhibitors may either increase or decrease extrasynaptic glycine levels, and this optimization may increase hypofunctional and decrease hyperfunctional NMDAR operation in various CNS disorders. Therefore, we suggest that GlyT1 inhibitors possess a more complex operation than just glycine uptake inhibition.
To the best of our knowledge, neither preclinical nor clinical studies have been carried out so far to elucidate the impact of GlyT1 inhibitors on the development of opioid analgesic tolerance. Large evidence exists regarding the efficacy of GlyT1 inhibitors in experimental schizophrenia models, though they have failed in phase III clinical studies. As mentioned above, the key player is NMDAR, which undergoes a hypofunctioning state in schizophrenia or hyperfunctioning state in opioid analgesic tolerance. As a limitation, glycine concentrations and their impact on spinal NMDARs to delay opioid analgesic tolerance necessitate the examination of GlyT1 inhibitors following acute and chronic administration. Yet, the safety of these drugs needs to be assessed under these protocols focusing on organ functions, particularly respiration and motor operation.
Author Contributions
Conceptualization, M.A.-K. and L.G.H.J.; methodology, Z.T.P.; software, A.R.G. and I.B.; validation, L.K.; formal analysis, A.R.G.; investigation, A.R.G. and Z.T.P.; data curation, A.R.G.; writing—original draft preparation, M.A.-K., L.G.H.J., A.R.G. and F.Z.; writing—review and editing, M.A.-K., L.G.H.J., L.K. and F.Z.; visualization, F.Z. and L.K.; supervision, M.A.-K., L.G.H.J., F.Z. and L.K.; project administration, A.R.G., F.Z. and M.A.-K.; funding acquisition, F.Z. and M.A.-K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, grant number FK_138389, and the Higher Education Institutional Excellence Programme of the Ministry of Human Capacities in Hungary, within the framework of the Neurology Thematic Programme of Semmelweis University (TKP 2021 EGA-25). A.R.G. was supported by “Semmelweis 250+ Kiválósági Ph.D. Ösztöndíj” (EFOP-3.6.3-VEKOP-16-2017-00009).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Essmat, N.; Karádi, D.; Zádor, F.; Király, K.; Fürst, S.; Al-Khrasani, M. Insights into the Current and Possible Future Use of Opioid Antagonists in Relation to Opioid-Induced Constipation and Dysbiosis. Molecules 2023, 28, 7766. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, K.; Caputi, F.F.; Hanuska, A.; Kató, E.; Balogh, M.; Köles, L.; Palmisano, M.; Riba, P.; Hosztafi, S.; Romualdi, P.; et al. A new potent analgesic agent with reduced liability to produce morphine tolerance. Brain Res. Bull. 2015, 117, 32–38. [Google Scholar] [CrossRef]
- Cahill, C.M.; Walwyn, W.; Taylor, A.M.; Pradhan, A.A.; Evans, C.J. Allostatic Mechanisms of Opioid Tolerance Beyond Desensitization and Downregulation. Trends Pharmacol. Sci. 2016, 37, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Kaplovitch, E.; Gomes, T.; Camacho, X.; Dhalla, I.A.; Mamdani, M.M.; Juurlink, D.N. Sex Differences in Dose Escalation and Overdose Death during Chronic Opioid Therapy: A Population-Based Cohort Study. PLoS ONE 2015, 10, e0134550. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.J.; Krebs, E.E.; Hudson, T.; Brown, J.; Li, C.; Martin, B.C. Impact of opioid dose escalation on the development of substance use disorders, accidents, self-inflicted injuries, opioid overdoses and alcohol and non-opioid drug-related overdoses: A retrospective cohort study. Addiction 2020, 115, 1098–1112. [Google Scholar] [CrossRef]
- Henry, S.G.; Wilsey, B.L.; Melnikow, J.; Iosif, A.-M. Dose Escalation During the First Year of Long-Term Opioid Therapy for Chronic Pain. Pain Med. 2015, 16, 733–744. [Google Scholar] [CrossRef]
- Chen, S.-R.; Pan, H.-L. Blocking μ opioid receptors in the spinal cord prevents the analgesic action by subsequent systemic opioids. Brain Res. 2006, 1081, 119–125. [Google Scholar] [CrossRef]
- Ballantyne, J.C.; Koob, G.F. Allostasis theory in opioid tolerance. Pain 2021, 162, 2315–2319. [Google Scholar] [CrossRef]
- Fürst, S.; Zádori, Z.S.; Zádor, F.; Király, K.; Balogh, M.; László, S.B.; Hutka, B.; Mohammadzadeh, A.; Calabrese, C.; Galambos, A.R.; et al. On the Role of Peripheral Sensory and Gut Mu Opioid Receptors: Peripheral Analgesia and Tolerance. Molecules 2020, 25, 2473. [Google Scholar] [CrossRef]
- Gong, K.; Bhargava, A.; Jasmin, L. GluN2B N-methyl-D-aspartate receptor and excitatory amino acid transporter 3 are upregulated in primary sensory neurons after 7 days of morphine administration in rats: Implication for opiate-induced hyperalgesia. Pain 2016, 157, 147–158. [Google Scholar] [CrossRef]
- Manning, B.H.; Mao, J.; Frenk, H.; Price, D.D.; Mayer, D.J. Continuous co-administration of dextromethorphan or MK-801 with morphine: Attenuation of morphine dependence and naloxone-reversible attenuation of morphine tolerance. Pain 1996, 67, 79–88. [Google Scholar] [CrossRef]
- Trujillo, K.A.; Akil, H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 1991, 251, 85–87. [Google Scholar] [CrossRef] [PubMed]
- van der Staay, F.J.; Rutten, K.; Erb, C.; Blokland, A. Effects of the cognition impairer MK-801 on learning and memory in mice and rats. Behav. Brain Res. 2011, 220, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.W.B.; Ho, R. Controversies of the Effect of Ketamine on Cognition. Front. Psychiatry 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ma, R.; Jin, Y.; Fang, J.; Du, J.; Shao, X.; Liang, Y.; Fang, J. Molecular mechanisms of opioid tolerance: From opioid receptors to inflammatory mediators (Review). Exp. Ther. Med. 2021, 22, 1004. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of µ-Opioid Receptors: Desensitization, Phosphorylation, Internalization, and Tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed]
- Lemel, L.; Lane, J.R.; Canals, M. GRKs as Key Modulators of Opioid Receptor Function. Cells 2020, 9, 2400. [Google Scholar] [CrossRef]
- Duarte, M.L.; Devi, L.A. Post-translational Modifications of Opioid Receptors. Trends Neurosci. 2020, 43, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Liu, N.; Gintzler, A.R. Phosphorylation of unique C-terminal sites of the mu-opioid receptor variants 1B2 and 1C1 influences their Gs association following chronic morphine. J. Neurochem. 2020, 152, 449–467. [Google Scholar] [CrossRef]
- Bailey, C.P.; Smith, F.L.; Kelly, E.; Dewey, W.L.; Henderson, G. How important is protein kinase C in μ-opioid receptor desensitization and morphine tolerance? Trends Pharmacol. Sci. 2006, 27, 558–565. [Google Scholar] [CrossRef]
- Allouche, S.; Noble, F.; Marie, N. Opioid receptor desensitization: Mechanisms and its link to tolerance. Front. Pharmacol. 2014, 5, 280. [Google Scholar] [CrossRef]
- Dang, V.C.; Christie, M.J. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012, 165, 1704–1716. [Google Scholar] [CrossRef]
- Deng, M.; Chen, S.; Chen, H.; Luo, Y.; Dong, Y.; Pan, H. Mitogen-activated protein kinase signaling mediates opioid-induced presynaptic NMDA receptor activation and analgesic tolerance. J. Neurochem. 2019, 148, 275–290. [Google Scholar] [CrossRef]
- Eulenburg, V.; Hülsmann, S. Synergistic Control of Transmitter Turnover at Glycinergic Synapses by GlyT1, GlyT2, and ASC-1. Int. J. Mol. Sci. 2022, 23, 2561. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bao, Y.; Zheng, H.; Qin, Y.; Hua, B. Can Src protein tyrosine kinase inhibitors be combined with opioid analgesics? Src and opioid-induced tolerance, hyperalgesia and addiction. Biomed. Pharmacother. 2021, 139, 111653. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; A Kemp, J.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Price, D.; Mayer, D. Thermal hyperalgesia in association with the development of morphine tolerance in rats: Roles of excitatory amino acid receptors and protein kinase C. J. Neurosci. 1994, 14, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-S.; Cherng, C.-H.; Luk, H.-N.; Ho, S.-T.; Tung, C.-S. Effects of NMDA receptor antagonists on inhibition of morphine tolerance in rats: Binding at μ-opioid receptors. Eur. J. Pharmacol. 1996, 297, 27–33. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Mathie, A.; Peters, J. G Protein-coupled receptors. Br. J. Pharmacol. 2011, 164, S5–S113. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Das, B.; Yao, A.Y.; Yan, R. Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J. Alzheimer’s Dis. 2020, 78, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Pin, J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef]
- Tamaru, Y.; Nomura, S.; Mizuno, N.; Shigemoto, R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: Differential location relative to pre- and postsynaptic sites. Neuroscience 2001, 106, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Driessen, A.K. Vagal Afferent Processing by the Paratrigeminal Nucleus. Front. Physiol. 2019, 10, 475415. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.; Coderre, T.J. Comparison of nociceptive effects produced by intrathecal administration of mGluR agonists. NeuroReport 1996, 7, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.; Coderre, T.J. The contribution of metabotropic glutamate receptors (mGluRs) to formalin-induced nociception. Pain 1996, 68, 255–263. [Google Scholar] [CrossRef]
- Osikowicz, M.; Mika, J.; Przewlocka, B. The glutamatergic system as a target for neuropathic pain relief. Exp. Physiol. 2013, 98, 372–384. [Google Scholar] [CrossRef]
- Chiechio, S. Modulation of Chronic Pain by Metabotropic Glutamate Receptors. Adv. Pharmacol. 2016, 75, 63–89. [Google Scholar] [CrossRef]
- Carlton, S.M.; Hargett, G.L. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J. Comp. Neurol. 2007, 501, 780–789. [Google Scholar] [CrossRef]
- Okubo, M.; Yamanaka, H.; Kobayashi, K.; Noguchi, K. Differential expression of mGluRs in rat spinal dorsal horns and their modulatory effects on nocifensive behaviors. Mol. Pain 2019, 15, 1744806919875026. [Google Scholar] [CrossRef]
- Richardson-Burns, S.M.; Haroutunian, V.; Davis, K.L.; Watson, S.J.; Meador-Woodruff, J.H. Metabotropic glutamate receptor mRNA expression in the schizophrenic thalamus. Biol. Psychiatry 2000, 47, 22–28. [Google Scholar] [CrossRef]
- Neto, F.L.; Schadrack, J.; Berthele, A.; Zieglgänsberger, W.; Tölle, T.R.; Castro-Lopes, J.M. Differential distribution of metabotropic glutamate receptor subtype mRNAs in the thalamus of the rat. Brain Res. 2000, 854, 93–105. [Google Scholar] [CrossRef]
- Muly, E.C.; Maddox, M.; Smith, Y. Distribution of mGluR1α and mGluR5 immunolabeling in primate prefrontal cortex. J. Comp. Neurol. 2003, 467, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.M.; McQuail, J.A.; Schwabe, M.R.; Burke, S.N.; Setlow, B.; Bizon, J.L. Age-Related Declines in Prefrontal Cortical Expression of Metabotropic Glutamate Receptors that Support Working Memory. eNeuro 2018, 5, e0164-18. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.; Reeve, A.; Bowes, M.; Winter, J.; Wotherspoon, G.; Davis, A.; Schmid, P.; Gasparini, F.; Kuhn, R.; Urban, L. mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurones mediate inflammatory hyperalgesia. Neuropharmacology 2001, 40, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Rizzonelli, P.; Paterlini, M.; Moretto, G.; Knöpfel, T.; Kuhn, R.; Memo, M.; Spano, P. mGluR5 metabotropic glutamate receptor distribution in rat and human spinal cord: A developmental study. Neurosci. Res. 1997, 28, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Azkue, J.J.; Knöpfel, T.; Kuhn, R.; Mateos, J.M.; Grandes, P. Distribution of the metabotropic glutamate receptor subtype mGluR5 in rat midbrain periaqueductal grey and relationship with ascending spinofugal afferents. Neurosci. Lett. 1997, 228, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, E.; Boccella, S.; Marabese, I.; Pierretti, G.; Guida, F.; Maione, S. The Cold Case of Metabotropic Glutamate Receptor 6: Unjust Detention in the Retina? Curr. Neuropharmacol. 2019, 18, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Govea, R.; Zhou, S.; Carlton, S. Group III metabotropic glutamate receptors and transient receptor potential vanilloid 1 co-localize and interact on nociceptors. Neuroscience 2012, 217, 130–139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marabese, I.; de Novellis, V.; Palazzo, E.; Mariani, L.; Siniscalco, D.; Rodella, L.; Rossi, F.; Maione, S. Differential roles of mGlu8 receptors in the regulation of glutamate and γ-aminobutyric acid release at periaqueductal grey level. Neuropharmacology 2005, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.W.; Wu, L.-J.; Shum, F.; Quan, J.; Zhuo, M. Cingulate NMDA NR2B receptors contribute to morphine-induced analgesic tolerance. Mol. Brain 2008, 1, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, M.; Shimoyama, N.; Inturrisi, C.E.; Elliott, K. Oral ketamine produces a dose-dependent CNS depression in the rat. Life Sci. 1996, 60, PL9–PL14. [Google Scholar] [CrossRef] [PubMed]
- Nakama-Kitamura, M. The N-Methyl-D-aspartate receptor antagonist dizocilpine inhibits associative antinociceptive tolerance to morphine in mice: Relation with memory. J. Pharmacol. Sci. 2005, 97, 75–82. [Google Scholar] [CrossRef]
- Laulin, J.-P.; Maurette, P.; Corcuff, J.-B.; Rivat, C.G.; Chauvin, M.; Simonnet, G. The Role of Ketamine in Preventing Fentanyl-Induced Hyperalgesia and Subsequent Acute Morphine Tolerance. Obstet. Anesth. Dig. 2002, 94, 1263–1269. [Google Scholar] [CrossRef]
- González, P.; Cabello, P.; Germany, A.; Norris, B.; Contreras, E. Decrease of tolerance to, and physical dependence on morphine by glutamate receptor antagonists. Eur. J. Pharmacol. 1997, 332, 257–262. [Google Scholar] [CrossRef]
- Elliott, K.; Minami, N.; Kolesnikov, Y.A.; Pasternak, G.W.; Inturrisi, C.E. The NMDA Receptor antagonists, LY274614 and MK-801, and the nitric oxide synthase inhibitor, NG-nitro-L-arginine, attenuate analgesic tolerance to the mu-opioid morphine but not to kappa opioids. Pain 1994, 56, 69–75. [Google Scholar] [CrossRef]
- Gutstein, H.B.; Trujillo, K.A. MK-801 inhibits the development of morphine tolerance at spinal sites. Brain Res. 1993, 626, 332–334. [Google Scholar] [CrossRef]
- Mendez, I.A.; Trujillo, K.A. NMDA receptor antagonists inhibit opiate antinociceptive tolerance and locomotor sensitization in rats. Psychopharmacology 2008, 196, 497–509. [Google Scholar] [CrossRef]
- Harris, L.D.; Regan, M.C.; Myers, S.J.; Nocilla, K.A.; Akins, N.S.; Tahirovic, Y.A.; Wilson, L.J.; Dingledine, R.; Furukawa, H.; Traynelis, S.F.; et al. Novel GluN2B-Selective NMDA Receptor Negative Allosteric Modulator Possesses Intrinsic Analgesic Properties and Enhances Analgesia of Morphine in a Rodent Tail Flick Pain Model. ACS Chem. Neurosci. 2023, 14, 917–935. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Laurie, D.; Seeburg, P. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci. 1994, 14 Pt 2, 3180–3194. [Google Scholar] [CrossRef]
- Tolle, T.; Berthele, A.; Zieglgansberger, W.; Seeburg, P.; Wisden, W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J. Neurosci. 1993, 13, 5009–5028. [Google Scholar] [CrossRef]
- Luque, J.; Bleuel, Z.; Malherbe, P.; Richards, J. Alternatively spliced isoforms of the N-methyl-d-aspartate receptor subunit 1 are differentially distributed within the rat spinal cord. Neuroscience 1994, 63, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Yung, K.K.L. Localization of glutamate receptors in dorsal horn of rat spinal cord. NeuroReport 1998, 9, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, A. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. J. Physiol. 2000, 523, 621–628. [Google Scholar] [CrossRef]
- Ma, Q.-P.; Hargreaves, R. Localization of N-methyl-d-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience 2000, 101, 699–707. [Google Scholar] [CrossRef]
- Wong, H.; Liu, X.; Matos, M.F.; Chan, S.F.; Pérez-Otaño, I.; Boysen, M.; Cui, J.; Nakanishi, N.; Trimmer, J.S.; Jones, E.G.; et al. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J. Comp. Neurol. 2002, 450, 303–317. [Google Scholar] [CrossRef]
- Wee, K.S.; Zhang, Y.; Khanna, S.; Low, C. Immunolocalization of NMDA receptor subunit NR3B in selected structures in the rat forebrain, cerebellum, and lumbar spinal cord. J. Comp. Neurol. 2008, 509, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Woo, D.H.; Bae, J.Y.; Nam, M.-H.; An, H.; Ju, Y.H.; Won, J.; Choi, J.H.; Hwang, E.M.; Han, K.-S.; Bae, Y.C.; et al. Activation of Astrocytic μ-opioid Receptor Elicits Fast Glutamate Release Through TREK-1-Containing K2P Channel in Hippocampal Astrocytes. Front. Cell. Neurosci. 2018, 12, 393968. [Google Scholar] [CrossRef]
- Cubelos, B. Localización del Transportador de Glicina GLYT1 en Sinapsis Glutamatérgicas y Caracterización de su Interacción con Proteínas Con Dominios PDZ. 2004. Available online: https://dialnet.unirioja.es/servlet/tesis?codigo=33450&info=resumen&idioma=SPA (accessed on 14 December 2023).
- Scimemi, A.; Fine, A.; Kullmann, D.M.; Rusakov, D.A. NR2B-Containing Receptors Mediate Cross Talk among Hippocampal Synapses. J. Neurosci. 2004, 24, 4767–4777. [Google Scholar] [CrossRef] [PubMed]
- Mony, L.; Kew, J.N.; Gunthorpe, M.J.; Paoletti, P. Allosteric modulators of NR2B-containing NMDA receptors: Molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–1317. [Google Scholar] [CrossRef] [PubMed]
- Harsing, L.G., Jr.; Juranyi, Z.; Gacsalyi, I.; Tapolcsanyi, P.; Czompa, A.; Matyus, P. Glycine Transporter Type-1 and its Inhibitors. Curr. Med. Chem. 2006, 13, 1017–1044. [Google Scholar] [CrossRef] [PubMed]
- Harsing, L.G.; Matyus, P. Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: The role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 2013, 93, 110–119. [Google Scholar] [CrossRef]
- Olivares, L.; Aragón, C.; Giménez, C.; Zafra, F. Analysis of the Transmembrane Topology of the Glycine Transporter GLYT1. J. Biol. Chem. 1997, 272, 1211–1217. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Functional ‘glial’ GLYT1 glycine transporters expressed in neurons. J. Neurochem. 2010, 114, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, C.L. Inhibition of Glycine Re-Uptake: A Potential Approach for Treating Pain by Augmenting Glycine-Mediated Spinal Neurotransmission and Blunting Central Nociceptive Signaling. Biomolecules 2021, 11, 864. [Google Scholar] [CrossRef] [PubMed]
- Łątka, K.; Bajda, M. Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters. Int. J. Mol. Sci. 2022, 23, 8050. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, A.; Lakatos, P.P.; Balogh, M.; Zádor, F.; Karádi, D.; Zádori, Z.S.; Király, K.; Galambos, A.R.; Barsi, S.; Riba, P.; et al. Pharmacological Evidence on Augmented Antiallodynia Following Systemic Co-Treatment with GlyT-1 and GlyT-2 Inhibitors in Rat Neuropathic Pain Model. Int. J. Mol. Sci. 2021, 22, 2479. [Google Scholar] [CrossRef]
- Peiser-Oliver, J.M.; Evans, S.; Adams, D.J.; Christie, M.J.; Vandenberg, R.J.; Mohammadi, S.A. Glycinergic Modulation of Pain in Behavioral Animal Models. Front. Pharmacol. 2022, 13, 860903. [Google Scholar] [CrossRef]
- Li, X.-H.; Miao, H.-H.; Zhuo, M. NMDA Receptor Dependent Long-term Potentiation in Chronic Pain. Neurochem. Res. 2018, 44, 531–538. [Google Scholar] [CrossRef]
- Toth, E.; Weiss, B.; Banay-Schwartz, M. Effect of glycine derivatives on behavioral changes induced by 3-mercaptopropionic acid or phencyclidine in mice. Res. Commun. Psychol. Psychiatry Behav. 1986, 11, 1–9. [Google Scholar]
- Harsing, L.; Zsilla, G.; Matyus, P.; Nagy, K.; Marko, B.; Gyarmati, Z.; Timar, J. Interactions between glycine transporter type 1 (GlyT-1) and some inhibitor molecules—Glycine transporter type 1 and its inhibitors (Review). Acta Physiol. Hung. 2012, 99, 1–17. [Google Scholar] [CrossRef]
- Zhang, H.X.; Hyrc, K.; Thio, L.L. The glycine transport inhibitor sarcosine is an NMDA receptor co-agonist that differs from glycine. J. Physiol. 2009, 587, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Carland, J.; Mansfield, R.; Ryan, R.; Vandenberg, R. Oleoyl-l-carnitine inhibits glycine transport by GlyT2. Br. J. Pharmacol. 2013, 168, 891–902. [Google Scholar] [CrossRef]
- Brown, A.; Carlyle, I.; Clark, J.; Hamilton, W.; Gibson, S.; McGarry, G.; McEachen, S.; Rae, D.; Thorn, S.; Walker, G. Discovery and SAR of Org 24598—A Selective Glycine Uptake Inhibitor. Bioorganic Med. Chem. Lett. 2001, 11, 2007–2009. [Google Scholar] [CrossRef] [PubMed]
- Herdon, H.J.; Godfrey, F.M.; Brown, A.M.; Coulton, S.; Evans, J.R.; Cairns, W.J. Pharmacological assessment of the role of the glycine transporter GlyT-1 in mediating high-affinity glycine uptake by rat cerebral cortex and cerebellum synaptosomes. Neuropharmacology 2001, 41, 88–96. [Google Scholar] [CrossRef]
- Harsing, L. An overview of Glyt-1 inhibitors under evaluation for the treatment of schizophrenia. Drugs Future 2013, 38, 555–568. [Google Scholar] [CrossRef]
- Gilfillan, R.; Kerr, J.; Walker, G.; Wishart, G. Glycine transporters and their inhibitors. Top. Med. Chem. 2009, 4, 223–247. [Google Scholar]
- McBain, C.J.; Kleckner, N.W.; Wyrick, S.; Dingledine, R. Structural requirements for activation of the glycine coagonist site of N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Mol. Pharmacol. 1989, 36, 556–565. [Google Scholar] [PubMed]
- Lee, M.-Y.; Lin, Y.-R.; Tu, Y.-S.; Tseng, Y.J.; Chan, M.-H.; Chen, H.-H. Effects of sarcosine and N, N-dimethylglycine on NMDA receptor-mediated excitatory field potentials. J. Biomed. Sci. 2017, 24, 18. [Google Scholar] [CrossRef] [PubMed]
- Werdehausen, R.; Kremer, D.; Brandenburger, T.; Schlösser, L.; Jadasz, J.; Küry, P.; Bauer, I.; Aragón, C.; Eulenburg, V.; Hermanns, H. Lidocaine Metabolites Inhibit Glycine Transporter 1. Anesthesiology 2012, 116, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Long, K.D.; Mastropaolo, J.; Rosse, R.B.; Manaye, K.F.; Deutsch, S.I. Modulatory effects of d-serine and sarcosine on NMDA receptor-mediated neurotransmission are apparent after stress in the genetically inbred BALB/c mouse strain. Brain Res. Bull. 2006, 69, 626–630. [Google Scholar] [CrossRef]
- Watson, G.B.; Bolanowski, M.A.; Baganoff, M.P.; Deppeler, C.L.; Lanthorn, T.H. d-Cycloserine acts as a partial agonist at the glycine modulatory site of the NMDA receptor expressed inXenopus oocytes. Brain Res. 1990, 510, 158–160. [Google Scholar] [CrossRef]
- Harrison, C.; Smart, D.; Lambert, D.G. Stimulatory effects of opioids. Br. J. Anaesth. 1998, 81, 20–28. [Google Scholar] [CrossRef]
- Corkrum, M.; Rothwell, P.E.; Thomas, M.J.; Kofuji, P.; Araque, A. Opioid-Mediated Astrocyte–Neuron Signaling in the Nucleus Accumbens. Cells 2019, 8, 586. [Google Scholar] [CrossRef] [PubMed]
- López-Corcuera, B.; Geerlings, A.; Aragón, C. Glycine neurotransmitter transporters: An update. Mol. Membr. Biol. 2001, 18, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Adams, R.; Betz, H.; Schloss, P. Modulation of a Recombinant Glycine Transporter (GLYT1b) by Activation of Protein Kinase C. J. Neurochem. 1995, 65, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Miller, G.M. A Receptor Mechanism for Methamphetamine Action in Dopamine Transporter Regulation in Brain. J. Pharmacol. Exp. Ther. 2009, 330, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.M. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J. Neurochem. 2011, 116, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Hanuska, A.; Szénási, G.; Albert, M.; Koles, L.; Varga, A.; Szabo, A.; Matyus, P.; Harsing, L.G. Some Operational Characteristics of Glycine Release in Rat Retina: The Role of Reverse Mode Operation of Glycine Transporter Type-1 (GlyT-1) in Ischemic Conditions. Neurochem. Res. 2016, 41, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, L.J.; Babey, A.-M. Synthesis of the Mechanisms of Opioid Tolerance: Do We Still Say NO? Cell. Mol. Neurobiol. 2021, 41, 927–948. [Google Scholar] [CrossRef]
- Ibuki, T.; Marsala, M.; Masuyama, T.; Yaksh, T.L. Spinal amino acid release and repeated withdrawal in spinal morphine tolerant rats. Br. J. Pharmacol. 2003, 138, 689–697. [Google Scholar] [CrossRef]
- Cortese, K.; Gagliani, M.C.; Raiteri, L. Interactions between Glycine and Glutamate through Activation of Their Transporters in Hippocampal Nerve Terminals. Biomedicines 2023, 11, 3152. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Siri, A.; Passalacqua, M.; Melloni, E.; Raiteri, M.; Bonanno, G. Glycine taken up through GLYT1 and GLYT2 heterotransporters into glutamatergic axon terminals of mouse spinal cord elicits release of glutamate by homotransporter reversal and through anion channels. Biochem. Pharmacol. 2005, 69, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-L.; Chen, S.-R.; Chen, H.; Pan, H.-L. Chronic Opioid Potentiates Presynaptic but Impairs Postsynaptic N-Methyl-d-aspartic Acid Receptor Activity in Spinal Cords: Implications for opioid hyperalgesia and tolerance. J. Biol. Chem. 2012, 287, 25073–25085. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. Mu-Opioid Receptors Transiently Activate the Akt-nNOS Pathway to Produce Sustained Potentiation of PKC-Mediated NMDAR-CaMKII Signaling. PLoS ONE 2010, 5, e11278. [Google Scholar] [CrossRef] [PubMed]
- Marques, B.L.; Oliveira-Lima, O.C.; Carvalho, G.A.; de Almeida Chiarelli, R.; Ribeiro, R.I.; Parreira, R.C.; da Madeira Freitas, E.M.; Resende, R.R.; Klempin, F.; Ulrich, H.; et al. Neurobiology of glycine transporters: From molecules to behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-J.; Lane, H.-Y.; Tsai, G.E. NMDA Pathology and Treatment of Schizophrenia. Curr. Pharm. Des. 2014, 20, 5118–5126. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Balla, A.; Sershen, H.; Lajtha, A. Reversal of phencyclidine-induced effects by glycine and glycine transport inhibitors. Biol. Psychiatry 1999, 45, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Adell, A. Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules 2020, 10, 947. [Google Scholar] [CrossRef]
- Pei, J.-C.; Luo, D.-Z.; Gau, S.-S.; Chang, C.-Y.; Lai, W.-S. Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients with Schizophrenia. Front. Psychiatry 2021, 12, 742058. [Google Scholar] [CrossRef]
- Rosenbrock, H.; Desch, M.; Wunderlich, G. Development of the novel GlyT1 inhibitor, iclepertin (BI 425809), for the treatment of cognitive impairment associated with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2023, 273, 1557–1566. [Google Scholar] [CrossRef]
- Dang, Y.-H.; Ma, X.-C.; Zhang, J.-C.; Ren, Q.; Wu, J.; Gao, C.-G.; Hashimoto, K. Targeting of NMDA Receptors in the Treatment of Major Depression. Curr. Pharm. Des. 2014, 20, 5151–5159. [Google Scholar] [CrossRef]
- Newell, D.W.; Barth, A.; Ricciardi, T.N.; Malouf, A.T. Glycine Causes Increased Excitability and Neurotoxicity by Activation of NMDA Receptors in the Hippocampus. Exp. Neurol. 1997, 145, 235–244. [Google Scholar] [CrossRef]
- Ghasemi, M.; Schachter, S.C. The NMDA receptor complex as a therapeutic target in epilepsy: A review. Epilepsy Behav. 2011, 22, 617–640. [Google Scholar] [CrossRef] [PubMed]
- Ugale, V.; Deshmukh, R.; Lokwani, D.; Reddy, P.N.; Khadse, S.; Chaudhari, P.; Kulkarni, P.P. GluN2B subunit selective N-methyl-D-aspartate receptor ligands: Democratizing recent progress to assist the development of novel neurotherapeutics. Mol. Divers. 2023, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Autry, A.E.; Tolias, K.F.; Krishnan, V. Ketamine: Neuroprotective or Neurotoxic? Front. Neurosci. 2021, 15, 672526. [Google Scholar] [CrossRef]
- Myslobodsky, M.; Golovchinsky, V.; Mintz, M. Ketamine: Convulsant or anti-convulsant? Pharmacol. Biochem. Behav. 1981, 14, 27–33. [Google Scholar] [CrossRef]
- Pinto, M.C.X.; Lima, I.V.d.A.; da Costa, F.L.P.; Rosa, D.V.; Mendes-Goulart, V.A.; Resende, R.R.; Romano-Silva, M.A.; de Oliveira, A.C.P.; Gomez, M.V.; Gomez, R.S. Glycine transporters type 1 inhibitor promotes brain preconditioning against NMDA-induced excitotoxicity. Neuropharmacology 2015, 89, 274–281. [Google Scholar] [CrossRef]
- Shen, H.-Y.; van Vliet, E.A.; Bright, K.-A.; Hanthorn, M.; Lytle, N.K.; Gorter, J.; Aronica, E.; Boison, D. Glycine transporter 1 is a target for the treatment of epilepsy. Neuropharmacology 2015, 99, 554–565. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).