

Synthesis, Cytotoxic, and Computational Screening of Some Novel Indole–1,2,4-Triazole-Based S-Alkylated N-Aryl Acetamides

 ,
,  ,
,  ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
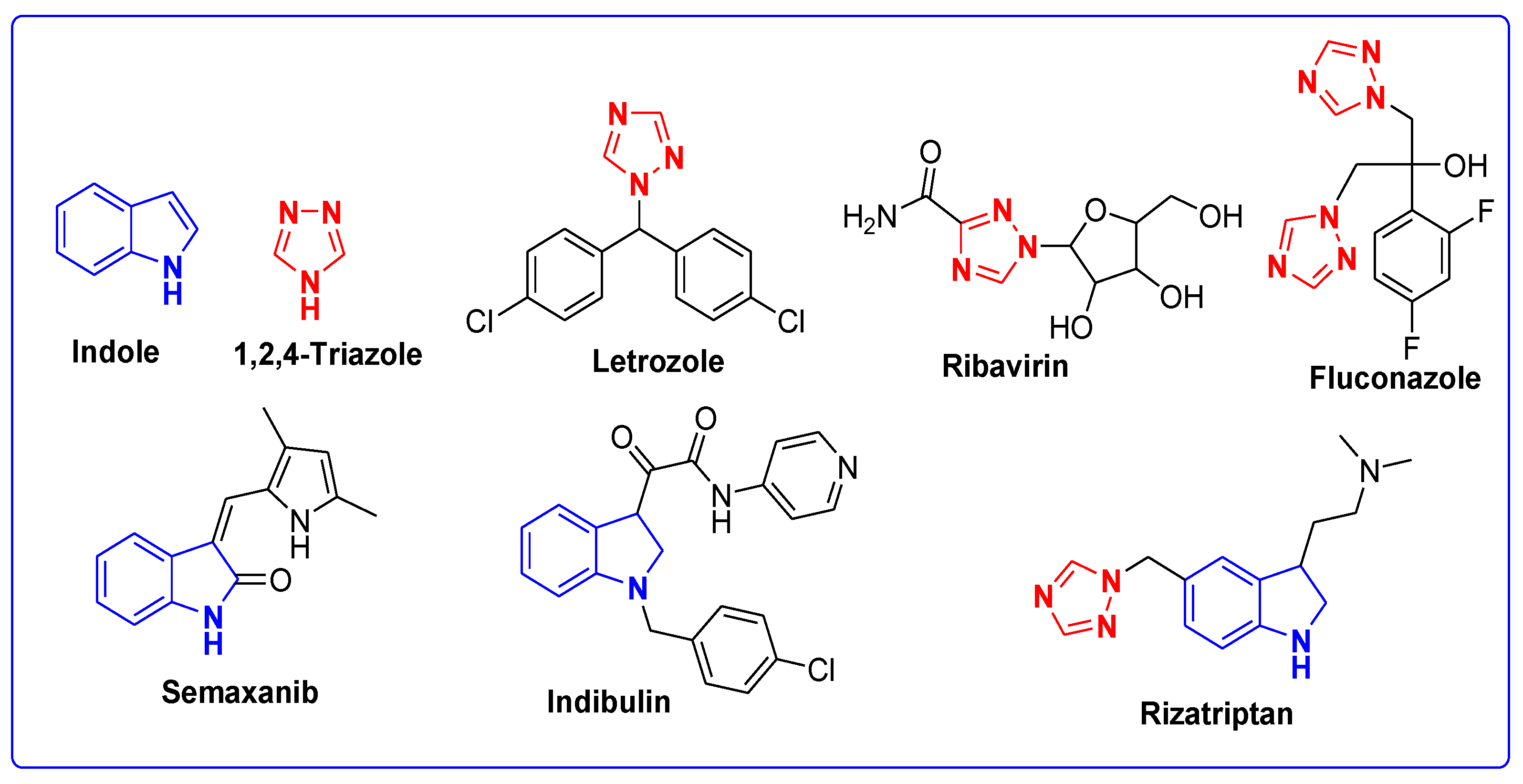
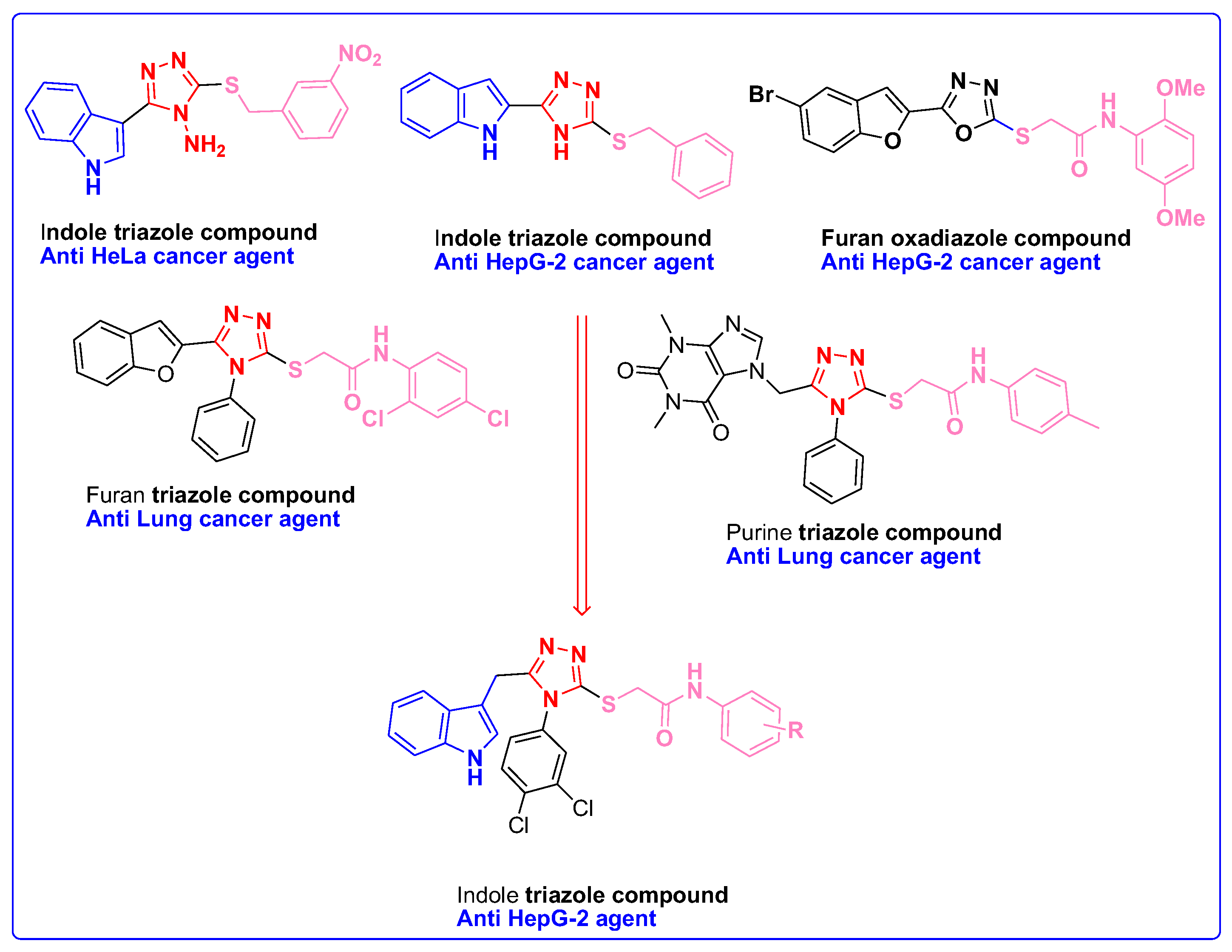
Molecular Fragmentation Hybridization Approach for Rationale Design
2. Materials and Methods
2.1. Materials for the Synthesis of Indole–1,2,4-Triazole Structural Hybrids
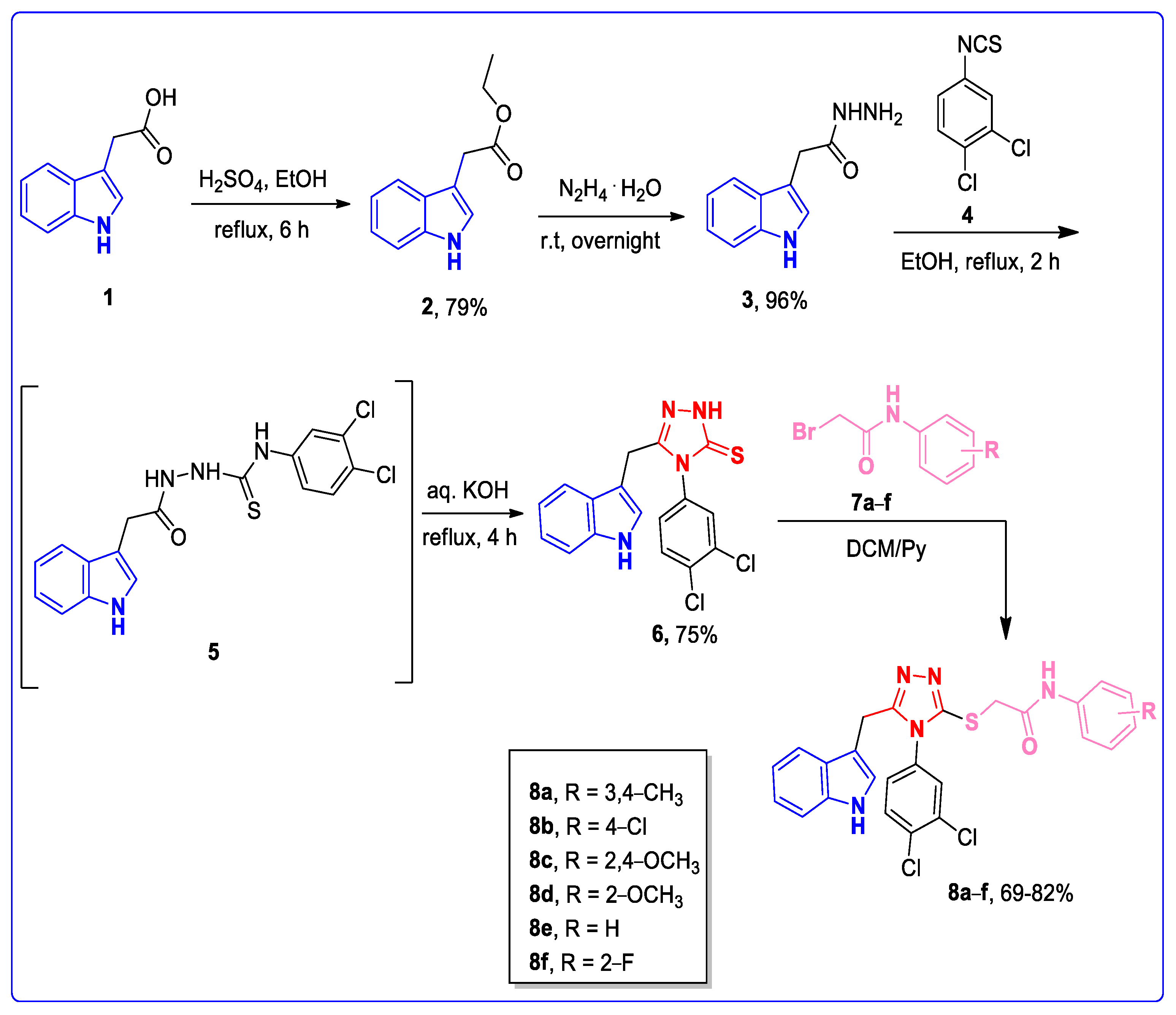
2.2. Synthetic Strategies and Procedures
2.2.1. Synthesis of Ethyl 2-(1H-indol-3-yl) Acetate 2
2.2.2. Synthesis of 2-(1H-indol-3-yl) Acetohydrazide 3
2.2.3. Synthesis of 5-((1H-indol-3-yl)methyl)-4-(3,4-dichlorophenyl)-4H-1,2,4-triazole-3-thiol 6
2.2.4. Synthetic Procedure to Synthesize Indole–1,2,4-Triazole Structural Hybrids 8a–f
2.2.5. Spectral Data of the Synthesized Compounds 8a–f
2.3. In Vitro Anticancer Activity
2.4. Molecular Docking of Indole–1,2,4-Triazoles
2.5. ADMET and Drug-Likeness Studies of Indole–1,2,4-Triazoles
2.6. MD Simulation of the Most Bioactive Indole–1,2,4-triazoles
2.7. MMPBSA/MMGBSA Binding Free Energies Estimation
2.8. DFT Studies
3. Results and Discussion
3.1. Chemistry
3.2. Spectral Interpretation of Compound 8b
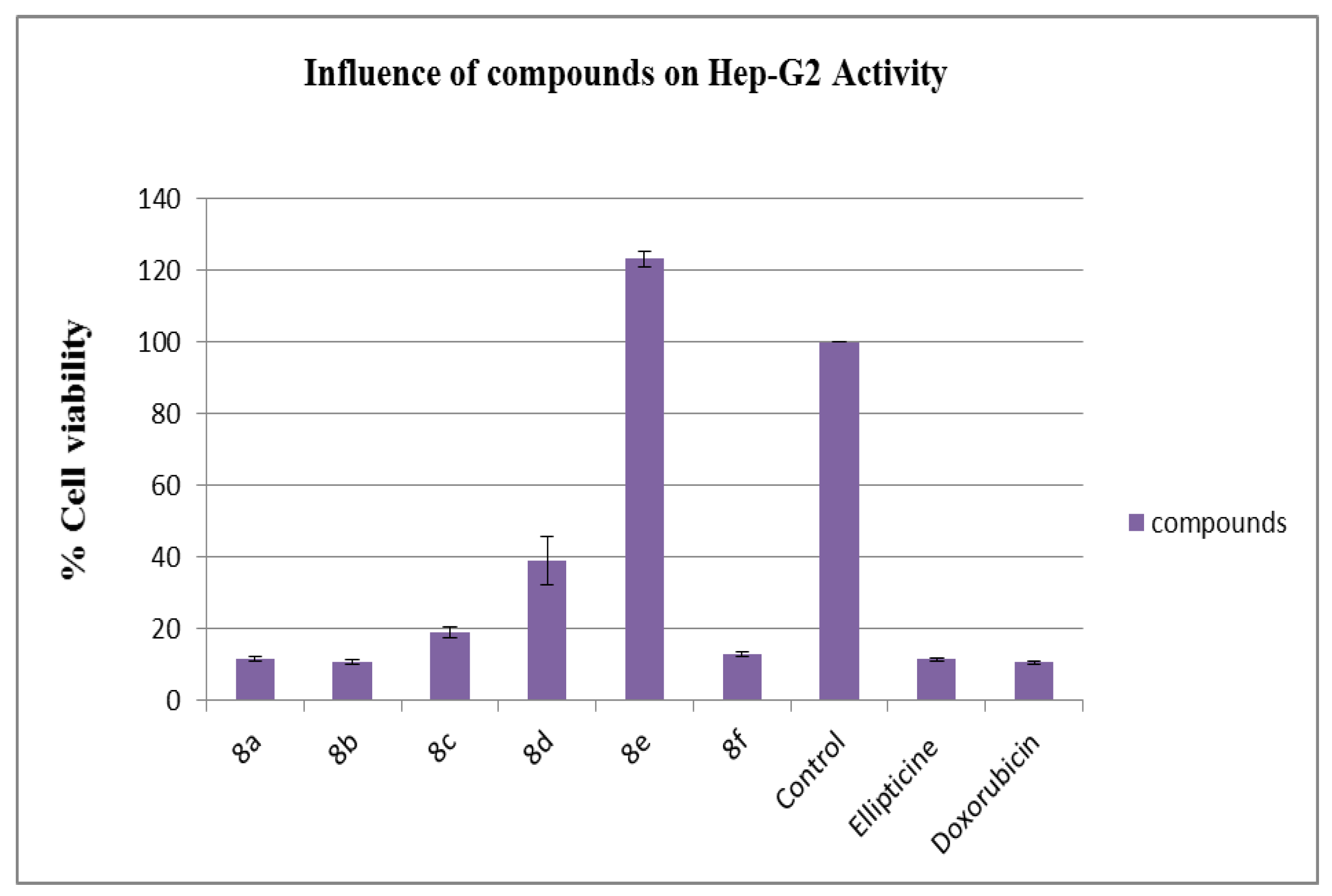
3.3. Anti-Hep-G2 Activity
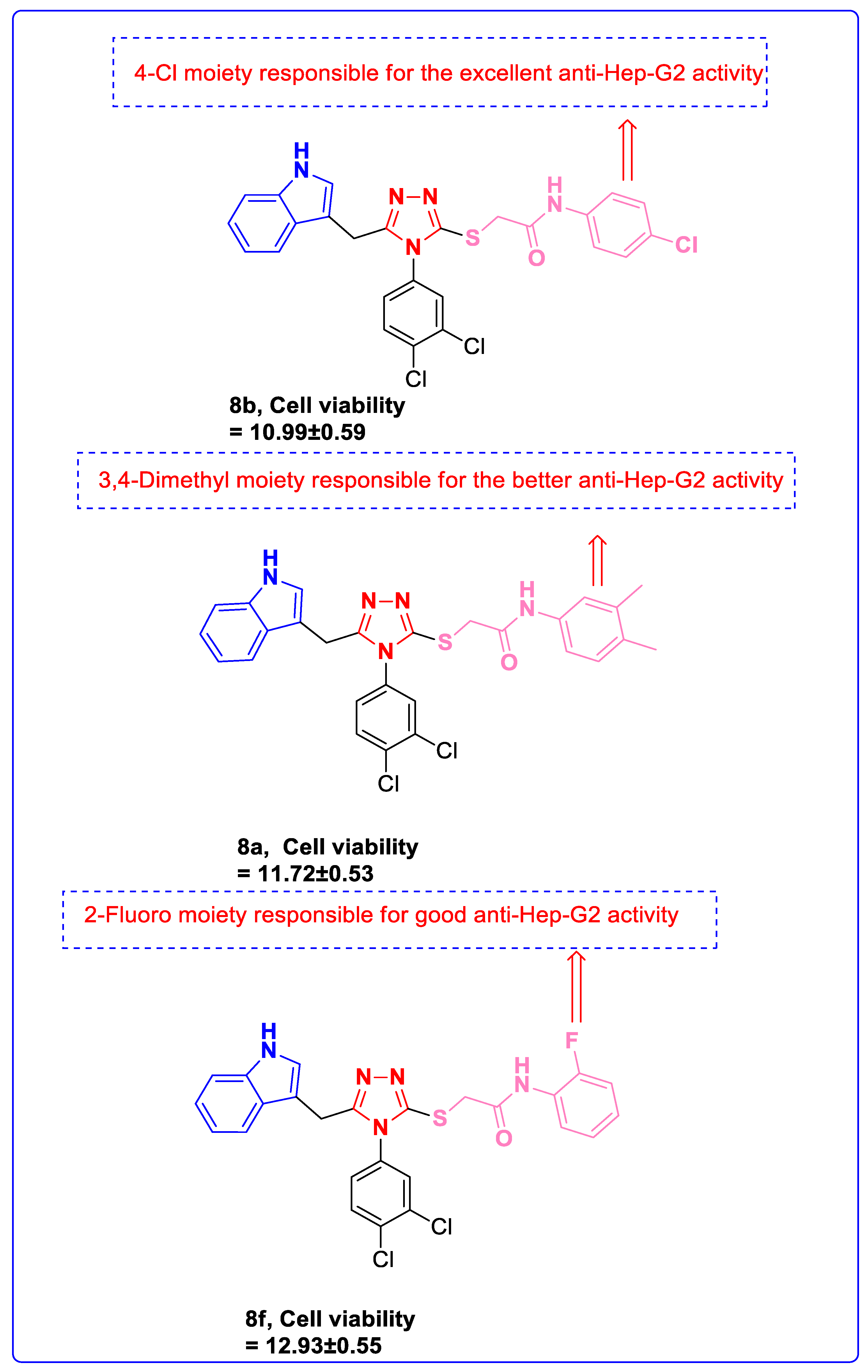
3.4. Structure–Activity Relationship (SAR)
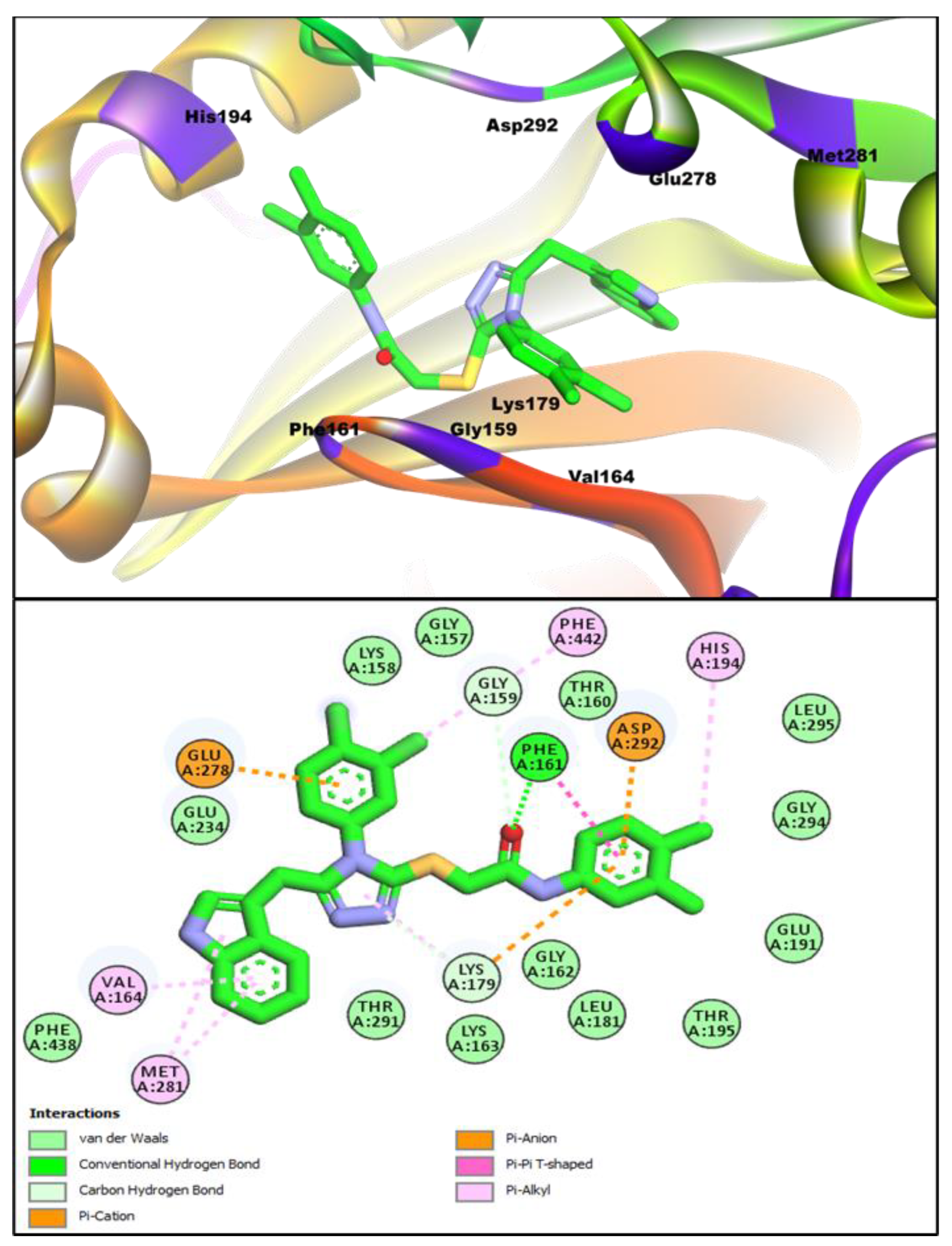
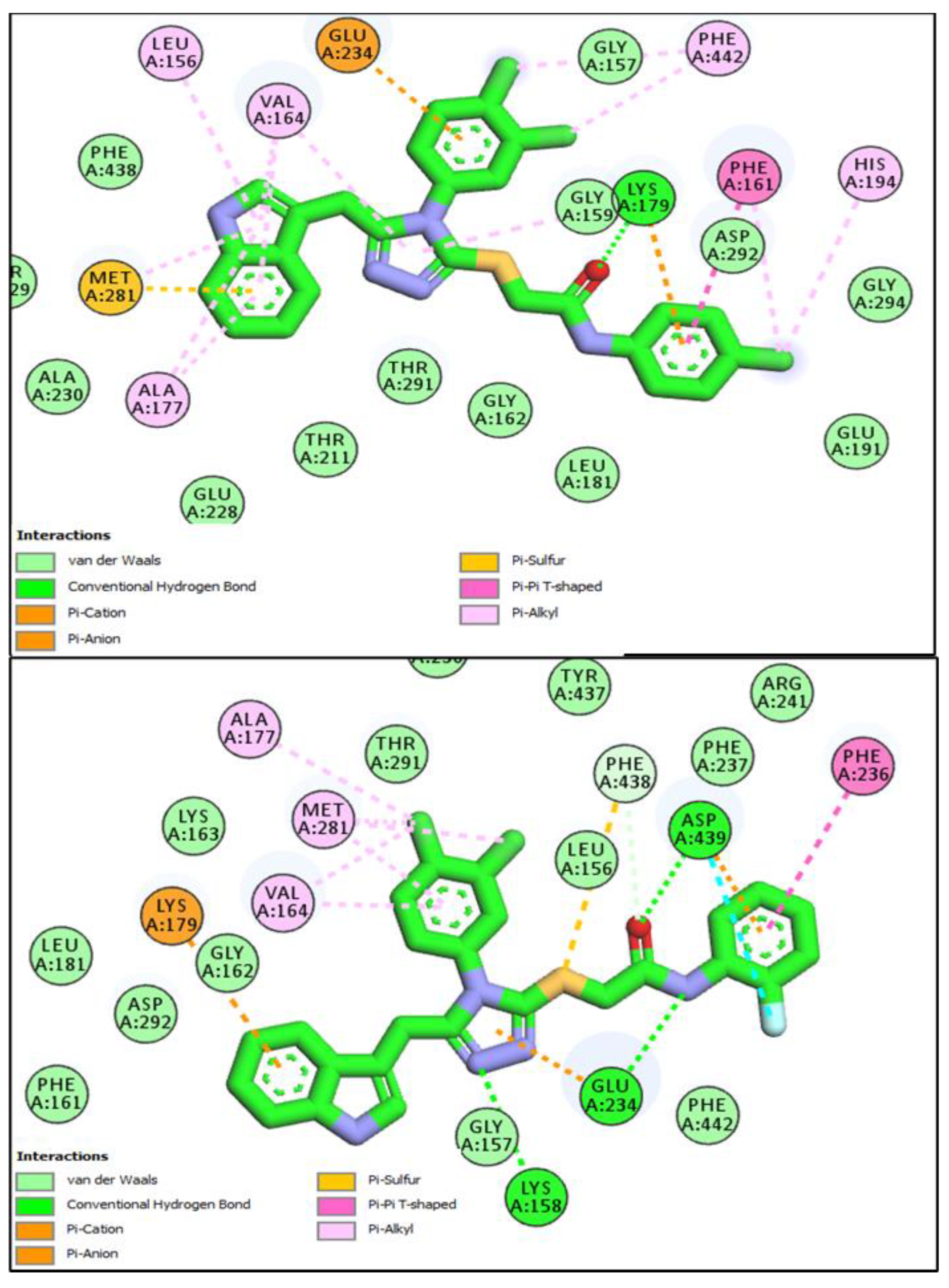
3.5. In Silico Molecular Docking Studies
3.6. In Silico ADMET Studies
3.7. In Silico Molecular Dynamic Simulations
3.7.1. Dynamic Structure Analysis
3.7.2. Binding Free Energies
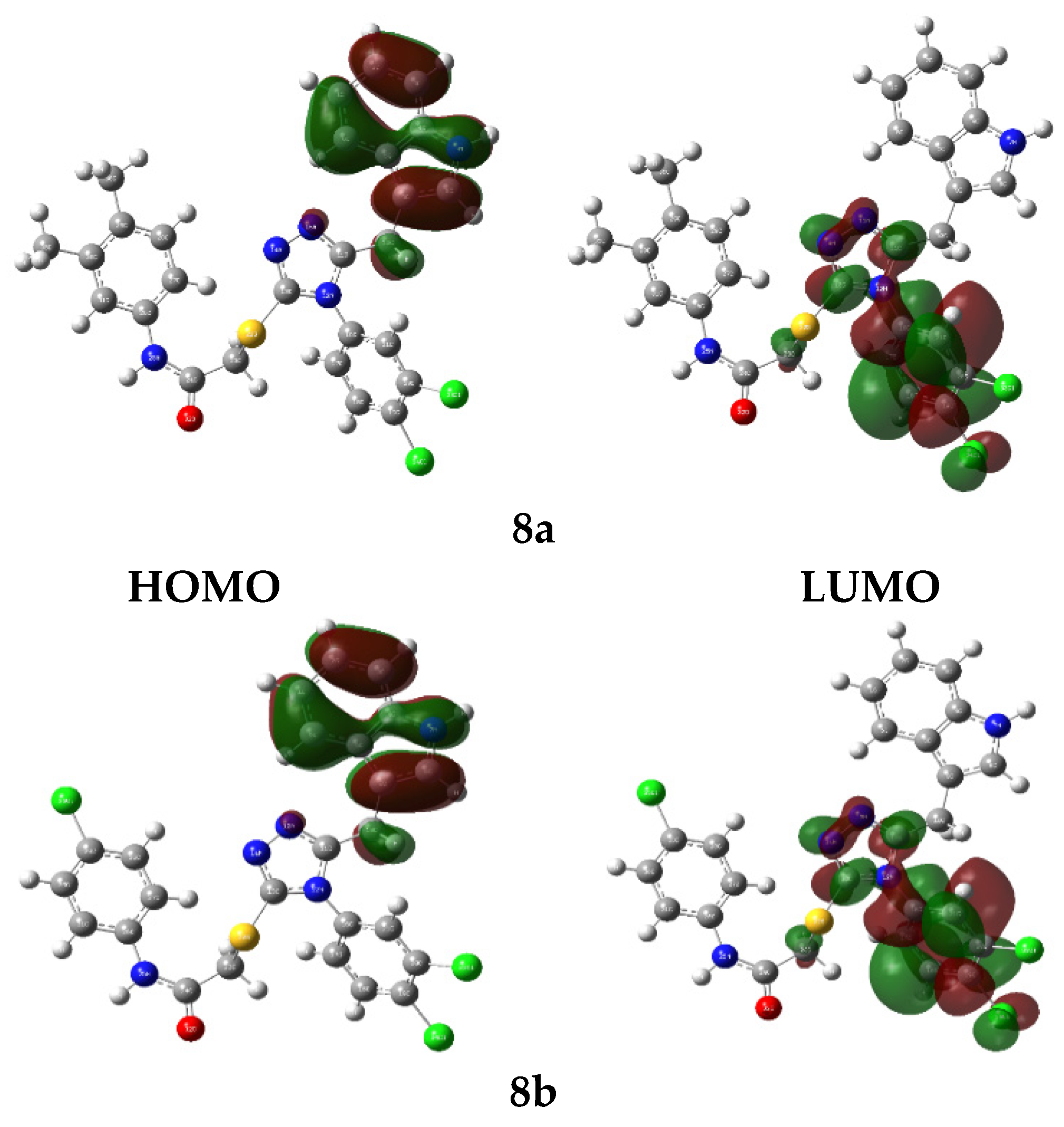
3.8. In Silico DFT Studies
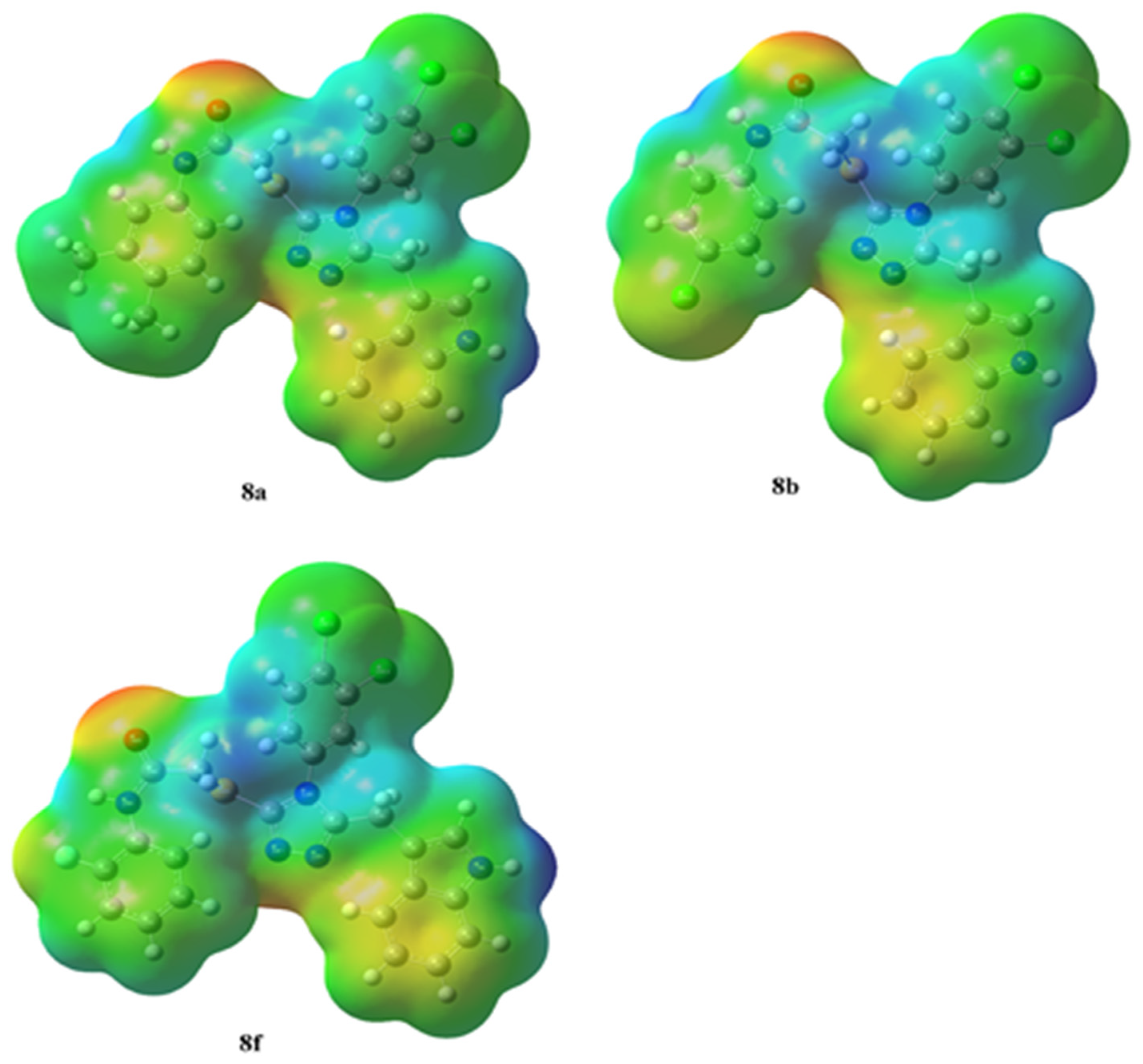
3.8.1. Molecular Electrostatic Potential Appraisal
3.8.2. HOMO-LUMO Evaluations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [Google Scholar] [CrossRef] [PubMed]
- El-Shershaby, M.H.; Ghiaty, A.; Bayoumi, A.H.; Ahmed, H.E.; El-Zoghbi, M.S.; El-Adl, K.; Abulkhair, H.S. 1, 2, 4-Triazolo [4,3-c] quinazolines: A bioisosterism-guided approach towards the development of novel PCAF inhibitors with potential anticancer activity. New J. Chem. 2021, 45, 11136–11152. [Google Scholar] [CrossRef]
- Abbas, S.Y.; Al-Harbi, R.A.; El-Sharief, M.A.S. Synthesis and anticancer activity of thiourea derivatives bearing a benzodioxole moiety with EGFR inhibitory activity, apoptosis assay and molecular docking study. Eur. J. Med. Chem. 2020, 198, 112363. [Google Scholar] [CrossRef] [PubMed]
- Koca, İ.; Özgür, A.; Coşkun, K.A.; Tutar, Y. Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorgan. Med. Chem. 2013, 21, 3859–3865. [Google Scholar] [CrossRef] [PubMed]
- Othman, E.M.; Fayed, E.A.; Husseiny, E.M.; Abulkhair, H.S. Apoptosis induction, PARP-1 inhibition, and cell cycle analysis of leukemia cancer cells treated with novel synthetic 1,2,3-triazole-chalcone conjugates. Bioorg. Chem. 2022, 123, 105762. [Google Scholar] [CrossRef] [PubMed]
- Mintz, K.J.; Leblanc, R.M. The use of nanotechnology to combat liver cancer: Progress and perspectives. BBA Rev. Cancer 2021, 1876, 188621. [Google Scholar] [CrossRef] [PubMed]
- Shewach, D.S.; Kuchta, R.D. Introduction to cancer chemotherapeutics. Chem. Rev. 2009, 109, 2859–2861. [Google Scholar] [CrossRef]
- İbiş, K.; Nalbat, E.; Çalışkan, B.; Kahraman, D.C.; Cetin-Atalay, R.; Banoglu, E. Synthesis and biological evaluation of novel isoxazole-piperazine hybrids as potential anti-cancer agents with inhibitory effect on liver cancer stem cells. Eur. J. Med. Chem. 2021, 221, 113489. [Google Scholar] [CrossRef]
- Man, S.; Luo, C.; Yan, M.; Zhao, G.; Ma, L.; Gao, W. Treatment for liver cancer: From sorafenib to natural products. Eur. J. Med. Chem. 2021, 224, 113690. [Google Scholar] [CrossRef]
- Güzelcan, E.A.; Baxendale, I.R.; Cetin-Atalay, R.; Baumann, M. Synthesis of new derivatives of boehmeriasin A and their biological evaluation in liver cancer. Eur. J. Med. Chem. 2019, 166, 243–255. [Google Scholar] [CrossRef]
- Khedr, F.; Ibrahim, M.K.; Eissa, I.H.; Abulkhair, H.S.; El-Adl, K. Phthalazine-based VEGFR-2 inhibitors: Rationale, design, synthesis, in silico, ADMET profile, docking, and anticancer evaluations. Arch. Pharm. 2021, 354, 2100201. [Google Scholar] [CrossRef] [PubMed]
- Nussbaumer, S.; Bonnabrya, P.; Veuthey, J.L.; Fleury-Souverain, S. Analysis of anticancer drugs: A review. Talanta 2011, 85, 2265–2289. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shuai, W.; Sun, H.; Xu, F.; Bi, Y.; Xu, J.; Ma, C.; Yao, H.; Zhu, Z.; Xu, S. Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur. J. Med. Chem. 2019, 163, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, R.; Zahoor, A.F.; Rasul, A.; Khan, S.G.; Ali, K.G. In-vitro cytotoxic evaluation of newly designed ciproflox-acinoxadiazole hybrids against human liver tumor cell line (Huh7). Pak. J. Pharm. Sci. 2021, 34, 1143–1148. [Google Scholar] [PubMed]
- Li, X.Y.; Shi, L.X.; Yao, X.M.; Jing, M.; Li, Q.-Q.; Wang, Y.-L.; Li, Q.-S. Functional vinorelbine plus schisandrin B liposomes destroying tumor metastasis in treatment of gastric cancer. Drug. Dev. Ind. Pharm. 2021, 47, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Turky, A.; Sherbiny, F.F.; Bayoumi, A.H.; Ahmed, H.E.; Abulkhair, H.S. Novel 1, 2, 4-triazole derivatives: Design, synthesis, anticancer evaluation, molecular docking, and pharmacokinetic profiling studies. Arch. Pharm. 2020, 353, 2000170. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- El-Shershaby, M.H.; El-Gamal, K.M.; Bayoumi, A.H.; El-Adl, K.; Ahmed, H.E.; Abulkhair, H.S. Synthesis, antimicrobial evaluation, DNA gyrase inhibition, and in silico pharmacokinetic studies of novel quinoline derivatives. Arch. Pharm. 2021, 354, 2000277. [Google Scholar] [CrossRef]
- Omar, A.M.; Ihmaid, S.; Habib, E.S.E.; Althagfan, S.S.; Ahmed, S.; Abulkhair, H.S.; Ahmed, H.E. The rational design, synthesis, and antimicrobial investigation of 2-amino-4-methylthiazole analogues inhibitors of GlcN-6-P synthase. Bioorg. Chem. 2020, 99, 103781. [Google Scholar] [CrossRef]
- Othman, E.M.; Fayed, E.A.; Husseiny, E.M.; Abulkhair, H.S. The effect of novel synthetic semicarbazone-and thiosemicarbazone-linked 1,2,3-triazoles on the apoptotic markers, VEGFR-2, and cell cycle of myeloid leukemia. Bioorg. Chem. 2022, 127, 105968. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Rashed, M.; Sharaky, M.; Abulkhair, H.S.; Hammouda, M.M.; Tawfik, H.O.; Shaldam, M.A. Novel fused imidazotriazines acting as promising top. II inhibitors and apoptotic inducers with greater selectivity against head and neck tumors: Design, synthesis, and biological assessments. Eur. J. Med. Chem. 2023, 259, 115661. [Google Scholar] [CrossRef]
- Baragaña, B.; Norcross, N.R.; Wilson, C.; Porzelle, A.; Hallyburton, I.; Grimaldi, R.; Osuna-Cabello, M.; Norval, S.; Riley, J.; Stojanovski, L.; et al. Discovery of a quinoline-4-carboxamide derivative with a novel mechanism of action, multistage antimalarial activity, and potent in vivo efficacy. J. Med. Chem. 2016, 59, 9672–9685. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; Patel, R.V.; Mahajan, D.H.; Parikh, P.A.; Mehta, G.N.; Pannecouque, C.; De Clercq, E.; Chikhalia, K.H. Different heterocycles functionalized S-triazine analogues: Design, synthesis and in vitro antimicrobial, antituberculosis, and anti-HIV assessment. J. Heterocycl. Chem. 2014, 51, 1641–1658. [Google Scholar] [CrossRef]
- Mohammad, B.D.; Baig, M.S.; Bhandari, N.; Siddiqui, F.A.; Khan, S.L.; Ahmad, Z.; Khan, F.S.; Tagde, P.; Jeandet, P. Heterocyclic compounds as dipeptidyl peptidase-IV inhibitors with special emphasis on oxadiazoles as potent anti-diabetic agents. Molecules 2022, 27, 6001. [Google Scholar] [CrossRef] [PubMed]
- Atukuri, D.; Gunjal, R.; Holagundi, N.; Korlahalli, B.; Gangannavar, S.; Akkasali, K. Contribution of N-heterocycles towards anti-tubercular drug discovery (2014–2019); predicted and reengineered molecular frameworks. Drug. Develop. Res. 2021, 82, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Nakhi, A.; Prasad, B.; Reddy, U.; Rao, R.M.; Sandra, S.; Kapavarapu, R.; Rambabu, D.; Krishna, G.R.; Reddy, C.M.; Ravada, K.; et al. A new route to indoles via in situ desilylation–Sonogashira strategy: Identification of novel small molecules as potential anti-tuberculosis agents. Med. Chem. Comm. 2011, 2, 1006–1010. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, N.; Chandra, R. The indole nucleus as a selective COX-2 inhibitor and anti-inflammatory agent (2011–2022). Org. Chem. Front. 2022, 9, 3624–3639. [Google Scholar] [CrossRef]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef]
- Sears, J.E.; Boger, D.L. Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure—function properties. Acc. Chem. Res. 2015, 48, 653–662. [Google Scholar] [CrossRef]
- Jagadeesan, S.; Karpagam, S. Novel series of N-acyl substituted indole based piperazine, thiazole and tetrazoles as potential antibacterial, anti-fungal, antioxidant and cytotoxic agents and their docking investigation as potential Mcl-1 inhibitors. J. Mol. Struct. 2023, 1271, 134013. [Google Scholar] [CrossRef]
- Aldawsari, H.M.; Singh, S.; Alhakamy, N.A.; Bakhaidar, R.B.; Halwani, A.A.; Badr-Eldin, S.M. Gum acacia functionalized colloidal gold nano-particles of letrozole as biocompatible drug delivery carrier for treatment of breast cancer. Pharmaceutics 2021, 13, 1554. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. In vitro dissolution of fluconazole and dipyridamole in gastrointestinal simulator (GIS), predicting in vivo dissolution and drug–drug interaction caused by acid-reducing agents. Mol. Pharm. 2015, 12, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsalam, S.; Sharaf-Eldin, M.; Soliman, S.; Elfert, A.; Badawi, R.; Ahmad, Y.K. Efficacy and safety of sofosbuvir plus ribavirin for treatment of cirrhotic patients with genotype 4 hepatitis C virus in real-life clinical practice. Arch. Virol. 2018, 163, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Shirinzadeh, H.; Ince, E.; Westwell, A.D.; Gurer-Orhan, H.; Suzen, S. Novel indole-based melatonin analogues substituted with triazole, thiadiazole and carbothioamides: Studies on their antioxidant, chemopreventive and cytotoxic activities. J. Enzym. Inhib. Med. Ch. 2016, 31, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, R.; Ziedan, N.; Ali, S.; El-Sadek, M.; Lashin, E.; Brancale, A.; Jones, A.J.; Westwell, A.D. Synthesis and evaluation of 3-(benzylthio)-5-(1H-indol-3-yl)-1,2,4-triazol-4-amines as Bcl-2 inhibitory anti-cancer agents. Bioorg. Med. Chem. Lett. 2013, 23, 2391–2394. [Google Scholar] [CrossRef]
- Boraei, A.T.; Gomaa, M.S.; El Sayed, H.; Duerkop, A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017, 125, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Zahoor, A.F.; Rasul, A.; Al-Hussain, S.A.; Faisal, S.; Ahmad, S.; Noor, R.; Muhammed, M.T.; Zaki, M.E.A. BTEAC catalyzed ultrasonic-assisted synthesis of bromobenzofuran-oxadiazoles: Unravelling anti-HepG-2 cancer therapeutic potential through in vitro and in silico studies. Int. J. Mol. Sci. 2023, 24, 3008. [Google Scholar] [CrossRef]
- Shahzadi, I.; Zahoor, A.F.; Rasul, A.; Mansha, A.; Ahmad, S.; Raza, Z. Synthesis, hemolytic studies, and in silico modeling of novel acefylline–1,2,4-triazole hybrids as potential anti-cancer agents against MCF-7 and A549. ACS Omega 2021, 6, 11943–11953. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the synergistic anticancer potential of benzofuran–oxadiazoles and triazoles: Improved ultrasound-and microwave-assisted synthesis, molecular docking, hemolytic, thrombolytic and anticancer evaluation of furan-based molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef]
- Saeed, S.; Zahoor, A.F.; Kamal, S.; Raza, Z.; Bhat, M.A. Unfolding the antibacterial activity and acetylcholinesterase inhibition potential of benzofuran-triazole hybrids: Synthesis, antibacterial, acetylcholinesterase inhibition, and molecular docking studies. Molecules 2023, 28, 6007. [Google Scholar] [CrossRef]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, M.E. Design, synthesis, antimicrobial evaluation, and laccase catalysis effect of novel benzofuran–oxadiazole and benzofuran–triazole hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Abreu, R.M.; Ferreira, I.C.; Calhelha, R.C.; Lima, R.T.; Vasconcelos, M.H.; Adega, F.; Chaves, R.; Queiroz, M.J. Anti-hepatocellular carcinoma activity using human HepG2 cells and hepatotoxicity of 6-substituted methyl 3-aminothieno[3,2-b]pyridine-2-carboxylate derivatives: In vitro evaluation, cell cycle analysis and QSAR studies. Eur. J. Med. Chem. 2011, 46, 5800–5806. [Google Scholar] [CrossRef] [PubMed]
- Al-Shafie, T.A.; Mahrous, E.A.; Shukry, M.; Alshahrani, M.Y.; Ibrahim, S.F.; Fericean, L.; Abdelkader, A.; Ali, M.A. A proposed association between improving energy metabolism of HepG2 cells by plant extracts and increasing their sensitivity to doxorubicin. Toxics 2023, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Rasul, A.; Di, J.; Millimouno, F.M.; Malhi, M.; Tsuji, I.; Ali, M.; Li, J.; Li, X. Reactive oxygen species mediate isoalantolactone-induced apoptosis in human prostate cancer cells. Molecules 2013, 18, 9382–9396. [Google Scholar] [CrossRef] [PubMed]
- Mills, N. ChemDraw Ultra 10.0 Cambridge Soft, 100 Cambridge Park Drive, Cambridge, MA 02140. www.Cambridgesoft.Com. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swisstargetprediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, F.-X.; Li, C.-Z.; Jia, C.-Y.; Su, S.-W.; Hao, G.-F.; Yang, G.-F. ACID: A free tool for drug repurposing using consensus inverse docking strategy. J. Cheminform. 2019, 11, 73. [Google Scholar] [CrossRef]
- Xu, Z.B.; Chaudhary, D.; Olland, S.; Wolfrom, S.; Czerwinski, R.; Malakian, K.; Lin, L.; Stahl, M.L.; Joseph-McCarthy, D.; Benander, C.; et al. Catalytic domain crystal structure of protein kinase C-θ (PKCθ). J. Biol. Chem. 2004, 279, 50401–50409. [Google Scholar] [CrossRef]
- Lin, K.; Lin, J.; Wu, W.-I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.A.; Morales, T.H.; Fried-man, L.S.; Skelton, N. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal. 2012, 5, ra37. [Google Scholar] [CrossRef]
- Heffron, T.P.; Heald, R.A.; Ndubaku, C.; Wei, B.; Augistin, M.; Do, S.; Wei, B.; Augistin, M.; Do, S.; Edgar, K.; et al. The rational design of selective benzoxazepin inhibitors of the α-isoform of phosphoinositide 3-kinase culminating in the identification of (S)-2-((2-(1-isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo [1,2-d][1,4]oxazepin-9-yl)oxy) propanamide (GDC-0326). J. Med. Chem. 2016, 59, 985–1002. [Google Scholar] [CrossRef]
- Oguro, Y.; Miyamoto, N.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; Miki, H.; Hori, A.; Ka-miyama, K.; Imamura, S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo [3,2-d] pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorgan. Med. Chem. 2010, 18, 7260–7273. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Molecular Operating Environment (MOE); 2015.10 Chemical Computing Group ULC: Montreal, QC, Canada, 2015. [Google Scholar]
- Vélizy-Villacoublay, F.S. Discovery Studio Modeling Environment, Release 3.5; Dassault Systèmes: Velizi Vilakuble, France, 2023. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. AD-METlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; Van, G.W. Molecular dynamics simulations. Curr. Opin. Struc. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB Force Field. AMBER 2014, 14, 29–31. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Kräutler, V.; Van, G.W.F.; Hünenberger, P.H. A fast shake algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Carugo, O. How root-mean-square distance (Rmsd) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Binding mode analysis, dynamic simulation and binding free energy calculations of the Mur F ligase from Acinetobacter baumannii. J. Mol. Graph. Model. 2017, 77, 72–85. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.J. XMGRACE, Version 5.1.19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian. J. Nat. Sci. Ed. 2009, 9, 53–54. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 5.0; Gaussian. Inc.: Wallingford, CT, USA, 2008. [Google Scholar]
- Bhullar, K.S.; Lagarón, N.O.; McGowa, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Biersack, B.; Li, Y.; Kong, D.; Bao, B.; Schobert, R.; Padhye, S.; Sarkar, F. Targeted regulation of PI3K/Akt/MTOR/NF-kB signaling by indole compounds and their derivatives: Mechanistic details and biological implications for cancer therapy. Anti-Cancer Agents Med. Chem. 2013, 13, 1002–1013. [Google Scholar] [CrossRef]
- Sarhan, A.A.M.; Boraei, A.T.A.; Barakat, A.; Nafie, M.S. Discovery of hydrazide-based pyri-dazino[4,5-b]indole scaffold as a new phosphoinositide 3-kinase (PI3K) inhibitor for breast cancer therapy. RSC Adv. 2020, 10, 19534–19541. [Google Scholar] [CrossRef]
- Mondal, D.; Amin, S.A.; Moinul, M.; Das, K.; Jha, T.; Gayen, S. How the structural properties of the indole derivatives are important in kinase targeted drug design?: A case study on tyrosine kinase inhibitors. Bioorgan. Med. Chem. 2022, 53, 116534. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Singh, V.; Shin, H.-S.; Patel, R.V. Kinase inhibitor indole derivatives as anticancer agents: A patent review. Recent Pat. Anti-Cancer Drug Discov. 2017, 12, 55–72. [Google Scholar] [CrossRef]
- Gao, G.R.; Li, M.Y.; Tong, L.J.; Wei, L.X.; Ding, J.; Xie, H.; Duan, W.H. Design, synthesis and biological evaluation of O-linked indoles as VEGFR-2 kinase inhibitors (I). Chin. Chem. Lett. 2015, 26, 1165–1168. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and subs. J. Chem. Res. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T. About the assignment of wave functions and eigenvalues to the individual electrons of an atom. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Ruiz-Morales, Y. HOMO-LUMO gap as an index of molecular size and structure for polycyclic aromatic hydrocarbons (PAHs) and asphaltenes: A theoretical study. I. J. Phys. Chem. A 2002, 106, 11283–11308. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HepG2 a% Cell Viability ± SD a |
|---|---|
| 8a | 11.72 ± 0.53 |
| 8b | 10.99 ± 0.59 |
| 8c | 18.92 ± 1.48 |
| 8d | 38.92 ± 6.75 |
| 8e | 123.21 ± 2.16 |
| 8f | 12.93 ± 0.55 |
| b Control | 100 ± 0.0 |
| Ellipticine [42] | 11.5 ± 0.55 |
| Doxorubicin [43] | 10.8 ± 0.41 |
| Entry | Chemical Structures | Binding Affinity with AKT1 Kcal/mol |
|---|---|---|
| 8a |  | −8.31 |
| 8b |  | −8.33 |
| 8f |  | −8.60 |
| Ipatasertib (control) |  | −8.25 |
| ADMET and Drug-Likeness Profile | 8a | 8b | 8f |
|---|---|---|---|
| TPSA | 75.6 Å | 75.6 Å | 75.6 Å |
| HIA | +ive | +ive | +ive |
| AMES toxic | Yes | Yes | Yes |
| MDCK cells‘ permeability | Low | Low | Low |
| Volume distribution (VD) | Optimal | Optimal | Optimal |
| Pfizer rule | Accepted | Accepted | Accepted |
| PAINS alerts | None | None | None |
| Chelator rule | Zero alerts | Zero alerts | Zero alerts |
| BBB penetration | Low | Low | Low |
| CYP3A4 substrate | Yes | Yes | Yes |
| Acute toxicity rule | Zero alerts | Zero alerts | Zero Alerts |
| Rat oral acute toxicity | Low | Low | Low |
| Clearance (CL) | 6.028 | 5.630 | 5.160 |
| Moderate | Moderate | Moderate |
| Compound | Energy Contribution | ||||||
|---|---|---|---|---|---|---|---|
| Electrostatic | Van der Waals | Gas Phase | Polar Solvation | Non-Polar Solvation | Solvation | Net | |
| AKT1-8a complex | −22.35 | −55.98 | −78.33 | 14.82 | −2.35 | 12.47 | −65.86 |
| AKT1-8b complex | −19.64 | −61.82 | −81.46 | 15.38 | −5.30 | 10.08 | −71.38 |
| AKT1-8f complex | −20.87 | −52.35 | −73.22 | 18.96 | −4.68 | −14.28 | −58.9 |
| Parameters | 8a | 8b | 8f |
|---|---|---|---|
| Etotal | −38,949.151 | −37,199.870 | −39,510.248 |
| EHOMO | −5.357 | −5.363 | −5.397 |
| ELUMO | −1.928 | −2.037 | −2.030 |
| ΔE | 3.429 | 3.326 | 3.367 |
| Ionization potential (IP= −EHOMO) | 5.357 | 5.363 | 5.397 |
| Electron affinity (A = −ELUMO) | 1.928 | 2.037 | 2.030 |
| Chemical potential (µ = −(I + A)/2) | −3.643 | −3.700 | −3.714 |
| Hardness (η = (I − A)/2) | 1.715 | 1.663 | 1.684 |
| Mulliken electronegativity (ᵡ = (I + A)/2) [79] | 3.643 | 3.700 | 3.714 |
| Softness (S = 1/2η) | 0.292 | 0.301 | 0.297 |
| Electrophilicity index (ꞷ = µ2/2η) [80] | 3.875 | 4.121 | 4.097 |
| Maximum charge transfer (ΔNmax = (I + A)/2(I − A)) [81] | 1.062 | 1.112 | 1.103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahoor, A.F.; Saeed, S.; Rasul, A.; Noreen, R.; Irfan, A.; Ahmad, S.; Faisal, S.; Al-Hussain, S.A.; Saeed, M.A.; Muhammed, M.T.; et al. Synthesis, Cytotoxic, and Computational Screening of Some Novel Indole–1,2,4-Triazole-Based S-Alkylated N-Aryl Acetamides. Biomedicines 2023, 11, 3078. https://doi.org/10.3390/biomedicines11113078
Zahoor AF, Saeed S, Rasul A, Noreen R, Irfan A, Ahmad S, Faisal S, Al-Hussain SA, Saeed MA, Muhammed MT, et al. Synthesis, Cytotoxic, and Computational Screening of Some Novel Indole–1,2,4-Triazole-Based S-Alkylated N-Aryl Acetamides. Biomedicines. 2023; 11(11):3078. https://doi.org/10.3390/biomedicines11113078
Chicago/Turabian StyleZahoor, Ameer Fawad, Sadaf Saeed, Azhar Rasul, Razia Noreen, Ali Irfan, Sajjad Ahmad, Shah Faisal, Sami A. Al-Hussain, Muhammad Athar Saeed, Muhammed Tilahun Muhammed, and et al. 2023. "Synthesis, Cytotoxic, and Computational Screening of Some Novel Indole–1,2,4-Triazole-Based S-Alkylated N-Aryl Acetamides" Biomedicines 11, no. 11: 3078. https://doi.org/10.3390/biomedicines11113078
APA StyleZahoor, A. F., Saeed, S., Rasul, A., Noreen, R., Irfan, A., Ahmad, S., Faisal, S., Al-Hussain, S. A., Saeed, M. A., Muhammed, M. T., Muhammad, Z. A., & Zaki, M. E. A. (2023). Synthesis, Cytotoxic, and Computational Screening of Some Novel Indole–1,2,4-Triazole-Based S-Alkylated N-Aryl Acetamides. Biomedicines, 11(11), 3078. https://doi.org/10.3390/biomedicines11113078