
Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders
 ,
,  , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Brief Overview of Distinct and Overlapping Clinical Features in Neurodegenerative Diseases
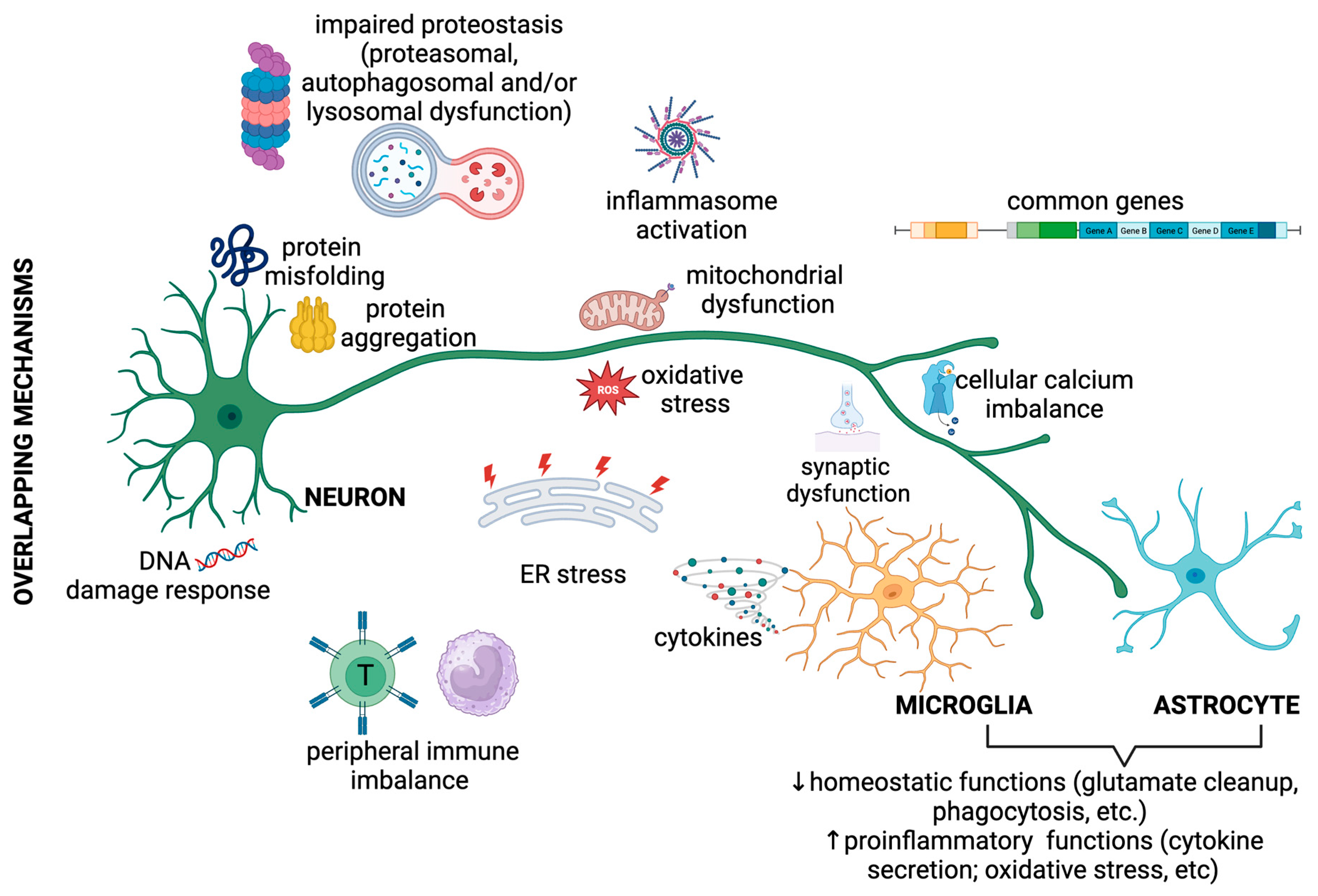
2. Overlapping Pathogenic Mechanisms in Neurodegenerative Diseases
2.1. Overlapping Proteinopathies
2.2. Overlapping Immune Imbalance
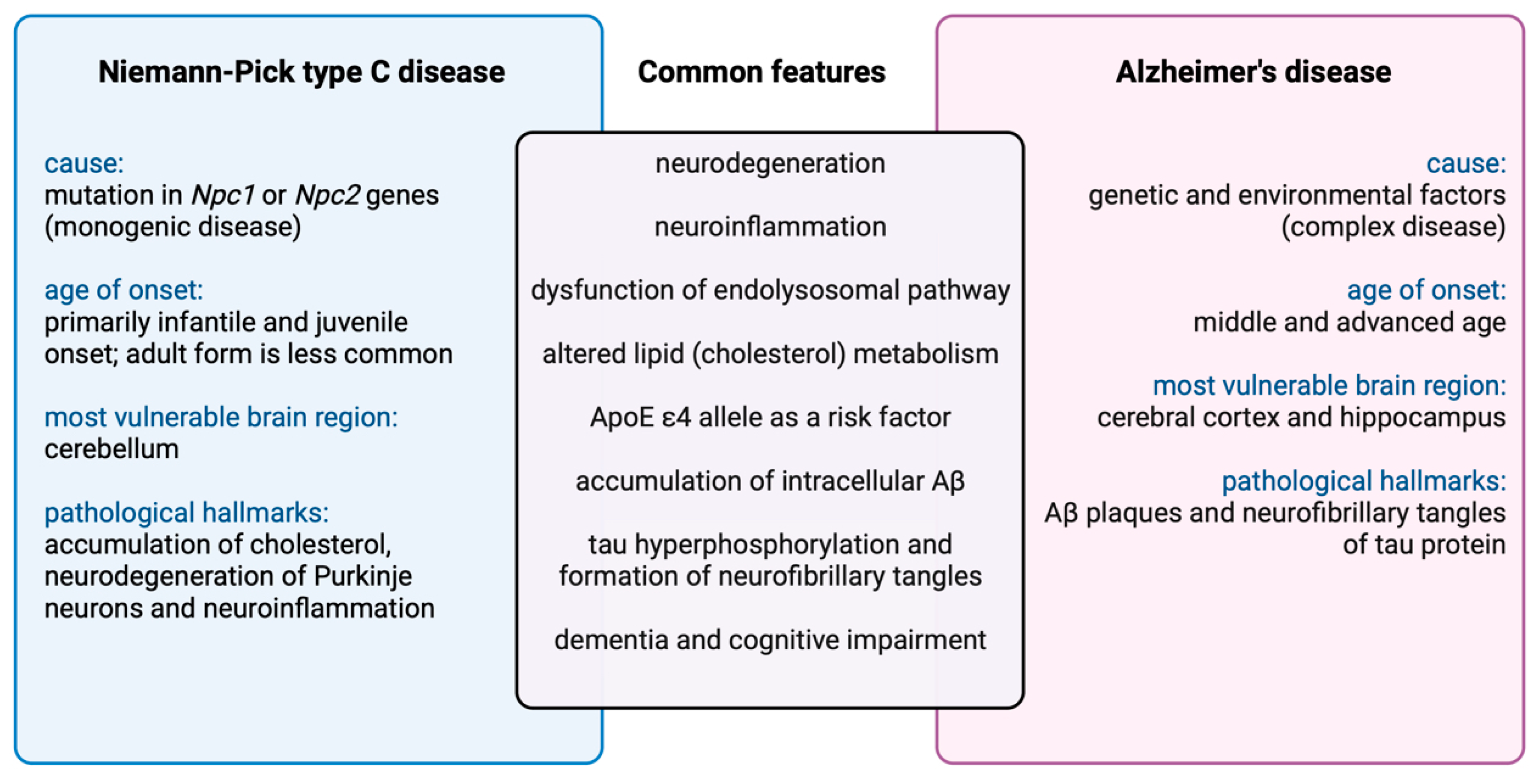
2.2.1. Immune Imbalance in Alzheimer’s Disease
2.2.2. Immune Imbalance in Niemann–Pick Type C Disease
2.2.3. Immune Imbalance in Parkinson’s Disease
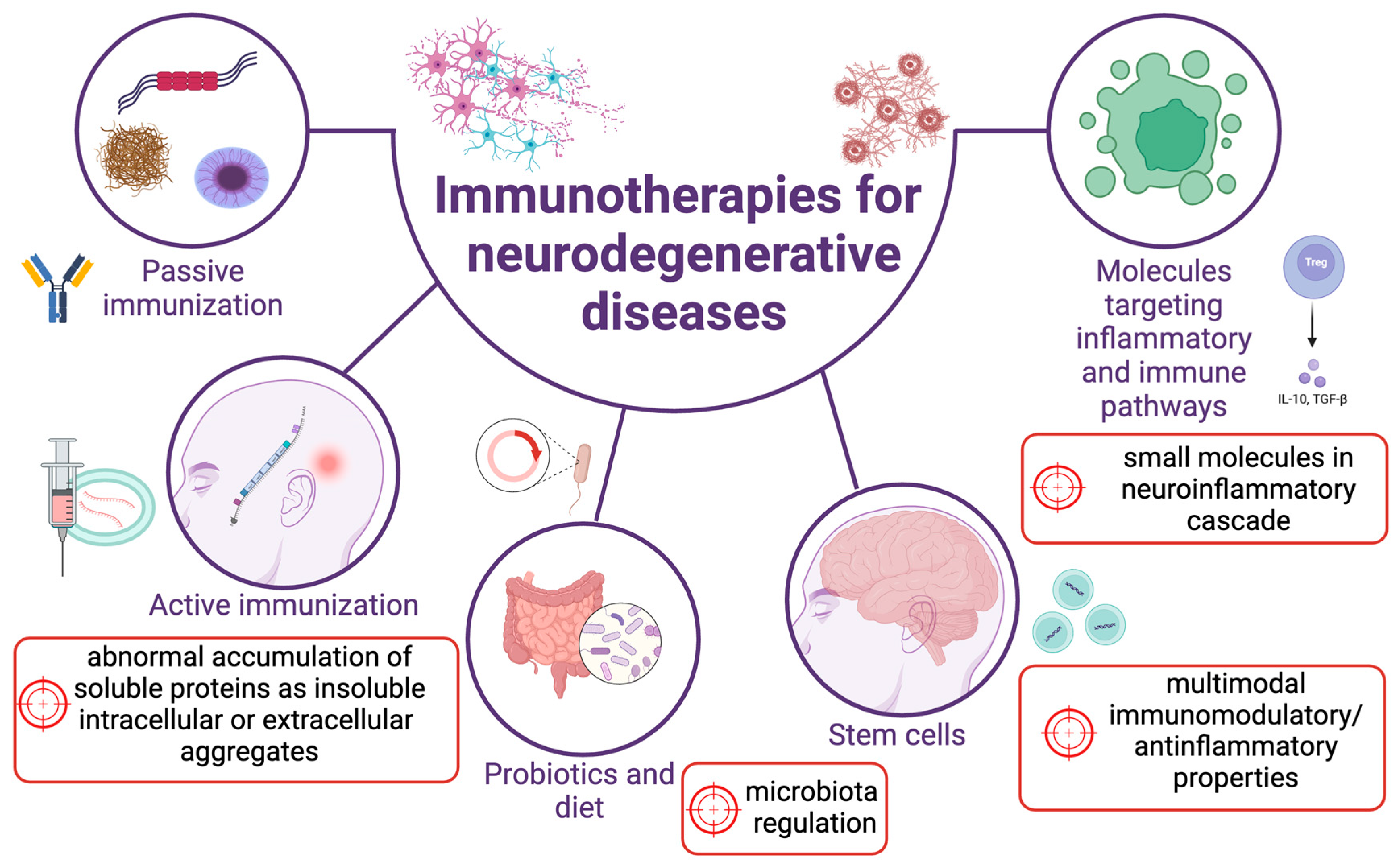
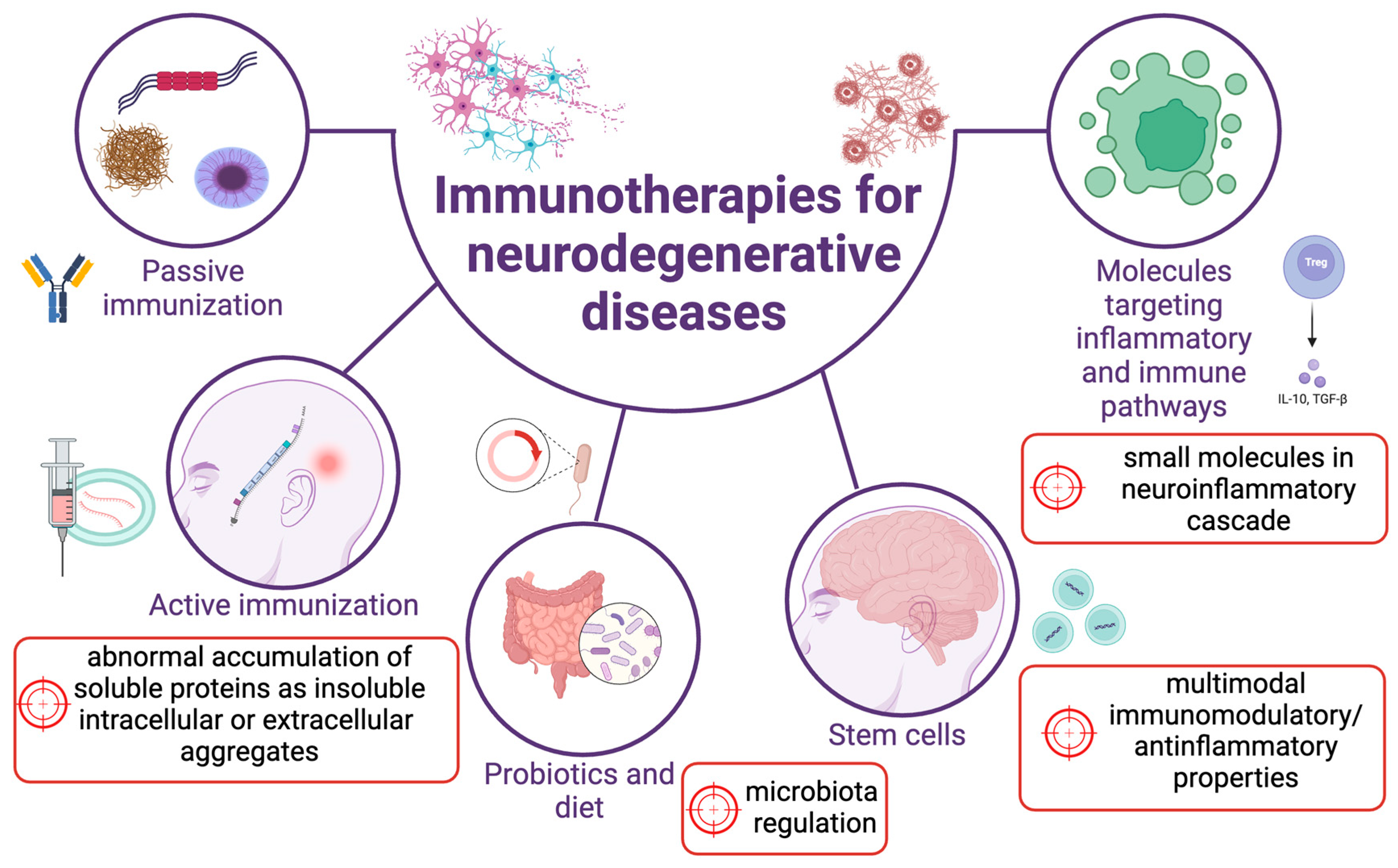
3. Common Therapeutic Approaches in Treating Immune (Dys)Function in Neurodegenerative Diseases
3.1. Passive and Active Vaccination Therapies Targeting Protein Aggregates
3.1.1. Clinical Trials on Aβ Immunisation
3.1.2. Clinical Trials on Tau Immunisation
3.1.3. Clinical Trials on α-syn Immunisation
3.1.4. Immunisation for ALS Treatment
3.2. Targeting Inflammatory Mediators
3.3. Targeting Complement
3.4. Stem Cell and Related Treatments
3.5. Targeting Microbiota
3.6. Potential Future Therapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwartz, M.; Deczkowska, A. Neurological Disease as a Failure of Brain–Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol. 2016, 37, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Appel, S.H. Immune Dysregulation in Amyotrophic Lateral Sclerosis: Mechanisms and Emerging Therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; de Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in Amyotrophic Lateral Sclerosis: Blurred Lines between Excessive Inflammation and Inefficient Immune Responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.-P. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Oksenberg, J.R.; Hauser, S.L. Multiple Sclerosis: Two Decades of Progress. Lancet Neurol. 2022, 21, 211–214. [Google Scholar] [CrossRef]
- Klotz, L.; Antel, J.; Kuhlmann, T. Inflammation in Multiple Sclerosis: Consequences for Remyelination and Disease Progression. Nat. Rev. Neurol. 2023, 19, 305–320. [Google Scholar] [CrossRef]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in Multiple Sclerosis. WIREs Mech. Dis. 2023, 15, e1583. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Liscic, R.M.; Storandt, M.; Cairns, N.J.; Morris, J.C. Clinical and Psychometric Distinction of Frontotemporal and Alzheimer Dementias. Arch. Neurol. 2007, 64, 535–540. [Google Scholar] [CrossRef]
- Mendez, M.F.; Selwood, A.; Mastri, A.R.; Frey, W.H. 2d Pick’s Disease versus Alzheimer’s Disease: A Comparison of Clinical Characteristics. Neurology 1993, 43, 289. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Lee, E.B.; Mackenzie, I.R. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv. Exp. Med. Biol. 2021, 1281, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Agosta, F.; Ferraro, P.M. Charting Frontotemporal Dementia: From Genes to Networks. J. Neuroimaging 2016, 26, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From Basic Research to the Clinic: Innovative Therapies for ALS and FTD in the Pipeline. Mol. Neurodegener. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.J.; Patterson, M.C.; Dambrosia, J.M.; Pikus, A.T.; Pentchev, P.G.; Sato, S.; Brady, R.O.; Barton, N.W. A Clinical Staging Classification for Type C Niemann-Pick Disease. Neurology 1992, 42, 2286. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann–Pick Diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar]
- Saito, Y.; Suzuki, K.; Nanba, E.; Yamamoto, T.; Ohno, K.; Murayama, S. Niemann–Pick Type C Disease: Accelerated Neurofibrillary Tangle Formation and Amyloid β Deposition Associated with Apolipoprotein E Ε4 Homozygosity. Ann. Neurol. 2002, 52, 351–355. [Google Scholar] [CrossRef]
- Distl, R.; Treiber-Held, S.; Albert, F.; Meske, V.; Harzer, K.; Ohm, T.G. Cholesterol Storage and Tau Pathology in Niemann–Pick Type C Disease in the Brain. J. Pathol. 2003, 200, 104–111. [Google Scholar] [CrossRef]
- Treiber-Held, S.; Distl, R.; Meske, V.; Albert, F.; Ohm, T.G. Spatial and Temporal Distribution of Intracellular Free Cholesterol in Brains of a Niemann–Pick Type C Mouse Model Showing Hyperphosphorylated Tau Protein. Implications for Alzheimer’s Disease. J. Pathol. 2003, 200, 95–103. [Google Scholar] [CrossRef]
- Platt, N.; Speak, A.O.; Colaco, A.; Gray, J.; Smith, D.A.; Williams, I.M.; Wallom, K.; Platt, F.M. Immune Dysfunction in Niemann-Pick Disease Type C. J. Neurochem. 2016, 136, 74–80. [Google Scholar] [CrossRef]
- Gratwicke, J.; Jahanshahi, M.; Foltynie, T. Parkinson’s Disease Dementia: A Neural Networks Perspective. Brain 2015, 138, 1454–1476. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray, C.K.; Weintraub, D. Parkinson Disease-Associated Cognitive Impairment (Primer). Nat. Rev. Dis. Primers 2021, 7, 47. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic Lateral Sclerosis: A Neurodegenerative Disorder Poised for Successful Therapeutic Translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 2021, 15, 652111. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Seeley, W.W.; Boxer, A.L.; Hillis, A.E.; Knopman, D.S.; Ljubenov, P.A.; Miller, B.; Piguet, O.; Rademakers, R.; Whitwell, J.L. Frontotemporal Lobar Degeneration. Nat. Rev. Dis. Primers 2023, 9, 40. [Google Scholar] [CrossRef]
- Öberg, M.; Fabrik, I.; Fabrikova, D.; Zehetner, N.; Härtlova, A. The Role of Innate Immunity and Inflammation in Parkinson’s Disease. Scand. J. Immunol. 2021, 93, e13022. [Google Scholar] [CrossRef]
- Perry, V.H. Innate Inflammation in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, A009373. [Google Scholar] [CrossRef]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and Inflammation in Parkinson’s Disease. Park. Relat. Disord. 2012, 18, S207–S209. [Google Scholar] [CrossRef]
- Standaert, D.G.; Harms, A.S.; Childers, G.M.; Webster, J.M. Disease Mechanisms as Subtypes: Inflammation in Parkinson Disease and Related Disorders. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2023; Volume 193, ISBN 0072-9752. [Google Scholar]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J. Serum Immune Markers and Disease Progression in an Incident Parkinson’s Disease Cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef]
- Berriat, F.; Lobsiger, C.S.; Boillée, S. The Contribution of the Peripheral Immune System to Neurodegeneration. Nat. Neurosci. 2023, 26, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Henderson, R.D.; Kepp, K.P.; Eisen, A. ALS/FTD: Evolution, Aging, and Cellular Metabolic Exhaustion. Front. Neurol. 2022, 13, 890203. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Zaikin, A.; Gordleeva, S.; Ivanchenko, M.; Bonifazi, F.; Storci, G.; Bonafè, M. Inflammaging 2018: An Update and a Model. Semin. Immunol. 2018, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Are Comorbidities Compatible with a Molecular Pathological Classification of Neurodegenerative Diseases? Curr. Opin. Neurol. 2019, 32, 279–291. [Google Scholar] [CrossRef]
- Polido, S.A.; Stuani, C.; Voigt, A.; Banik, P.; Kamps, J.; Bader, V.; Grover, P.; Krause, L.J.; Zerr, I.; Matschke, J. Cross-Seeding by Prion Protein Inactivates TDP-43. Brain 2023, awad289, Online ahead of print. [Google Scholar] [CrossRef]
- Rus, T.; Mlakar, J.; Jamšek, J.; Trošt, M. Metabolic Brain Changes Can Predict the Underlying Pathology in Neurodegenerative Brain Disorders: A Case Report of Sporadic Creutzfeldt–Jakob Disease with Concomitant Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 13081. [Google Scholar] [CrossRef]
- Beach, T.G.; Malek-Ahmadi, M. Alzheimer’s Disease Neuropathological Comorbidities Are Common in the Younger-Old. J. Alzheimer’s Dis. 2021, 79, 389–400. [Google Scholar] [CrossRef]
- Riku, Y.; Yoshida, M.; Iwasaki, Y.; Sobue, G.; Katsuno, M.; Ishigaki, S. TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? Int. J. Mol. Sci. 2022, 23, 15755. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Petrucelli, L.; Liesinger, A.M.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W. Updated TDP-43 in Alzheimer’s Disease Staging Scheme. Acta Neuropathol. 2016, 131, 571–585. [Google Scholar] [CrossRef]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-Synuclein and Tau: Teammates in Neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-Synuclein in the Pathophysiology of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by α-Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.; Hasegawa, M. α-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef]
- De Santis, R.; Alfano, V.; de Turris, V.; Colantoni, A.; Santini, L.; Garone, M.G.; Antonacci, G.; Peruzzi, G.; Sudria-Lopez, E.; Wyler, E. Mutant FUS and ELAVL4 (HuD) Aberrant Crosstalk in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 27, 3818–3831. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS Disease Protein TDP-43 Is Actively Transported in Motor Neuron Axons and Regulates Axon Outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef]
- Šušnjar, U.; Škrabar, N.; Brown, A.-L.; Abbassi, Y.; Phatnani, H.; Cortese, A.; Cereda, C.; Bugiardini, E.; Cardani, R. Cell Environment Shapes TDP-43 Function with Implications in Neuronal and Muscle Disease. Commun. Biol. 2022, 5, 314. [Google Scholar] [CrossRef]
- Scheres, S.H.W.; Ryskeldi-Falcon, B.; Goedert, M. Molecular Pathology of Neurodegenerative Diseases by Cryo-EM of Amyloids. Nature 2023, 621, 701–710. [Google Scholar] [CrossRef]
- Shih, Y.-H.; Tu, L.-H.; Chang, T.-Y.; Ganesan, K.; Chang, W.-W.; Chang, P.-S.; Fang, Y.-S.; Lin, Y.-T.; Jin, L.-W.; Chen, Y.-R. TDP-43 Interacts with Amyloid-β, Inhibits Fibrillization, and Worsens Pathology in a Model of Alzheimer’s Disease. Nat. Commun. 2020, 11, 5950. [Google Scholar] [CrossRef]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-Terminal Domain of TDP-43 and α-Synuclein Interact Synergistically to Generate Neurotoxic Hybrid Fibrils. J. Mol. Biol. 2021, 433, 166953. [Google Scholar] [CrossRef]
- Deshaies, J.-E.; Shkreta, L.; Moszczynski, A.J.; Sidibé, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J. TDP-43 Regulates the Alternative Splicing of HnRNP A1 to Yield an Aggregation-Prone Variant in Amyotrophic Lateral Sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Čerček, U.; Yamoah, A.; Tripathi, P.; Aronica, E.; Goswami, A.; Rogelj, B. Abl Kinase-Mediated FUS Tyr526 Phosphorylation Alters Nucleocytoplasmic FUS Localization in FTLD-FUS. Brain 2023, 146, 4088–4104. [Google Scholar] [CrossRef]
- Cracco, L.; Doud, E.H.; Hallinan, G.I.; Garringer, H.J.; Jacobsen, M.H.; Richardson, R.M.; Buratti, E.; Vidal, R.; Ghetti, B.; Newell, K.L. Distinguishing Post-translational Modifications in Dominantly Inherited Frontotemporal Dementias: FTLD-TDP Type A (GRN) vs Type B (C9orf72). Neuropathol. Appl. Neurobiol. 2022, 48, e12836. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Rogelj, B. The Role of C-Abl Tyrosine Kinase in Brain and Its Pathologies. Cells 2023, 12, 2041. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R. Revisiting Protein Aggregation as Pathogenic in Sporadic Parkinson and Alzheimer Diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, M.; Funayama, M.; Matsuura, E.; Yoshino, H.; Li, Y.; Tsuyama, S.; Takashima, H.; Nishioka, K.; Hattori, N. Isolated Nigral Degeneration without Pathological Protein Aggregation in Autopsied Brains with LRRK2 p. R1441H Homozygous and Heterozygous Mutations. Acta Neuropathol. Commun. 2018, 6, 105. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of Genetic Synucleinopathies with Parkinsonism: Review of the Literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J. ER–Mitochondria Associations Are Regulated by the VAPB–PTPIP51 Interaction and Are Disrupted by ALS/FTD-Associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-Length TDP-43 and Its C-Terminal Fragments Activate Mitophagy in NSC34 Cell Line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Wang, S.; Li, H.; Zhang, L.; Zhang, H.; Wang, P.-H.; Zheng, X.; Yu, X.-F.; Wei, W. The SARS-CoV-2 Main Protease Induces Neurotoxic TDP-43 Cleavage and Aggregates. Signal Transduct. Target. Ther. 2023, 8, 109. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D. TDP-43 Triggers Mitochondrial DNA Release via MPTP to Activate CGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Su, C.; Zhao, K.; Xia, H.; Xu, Y. Peripheral Inflammatory Biomarkers in Alzheimer’s Disease and Mild Cognitive Impairment: A Systematic Review and Meta-analysis. Psychogeriatrics 2019, 19, 300–309. [Google Scholar] [CrossRef]
- Abdi, I.Y.; Ghanem, S.S.; El-Agnaf, O.M. Immune-Related Biomarkers for Parkinson’s Disease. Neurobiol. Dis. 2022, 170, 105771. [Google Scholar] [CrossRef]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central Nervous System Macrophages in Progressive Multiple Sclerosis: Relationship to Neurodegeneration and Therapeutics. J. Neuroinflammation 2022, 19, 45. [Google Scholar] [CrossRef]
- Acosta-Galeana, I.; Hernández-Martínez, R.; Reyes-Cruz, T.; Chiquete, E.; de Jesus Aceves-Buendia, J. RNA-Binding Proteins as a Common Ground for Neurodegeneration and Inflammation in Amyotrophic Lateral Sclerosis and Multiple Sclerosis. Front. Mol. Neurosci. 2023, 16, 1193636. [Google Scholar] [CrossRef]
- De Marchi, F.; Tondo, G.; Corrado, L.; Menegon, F.; Aprile, D.; Anselmi, M.; D’Alfonso, S.; Comi, C.; Mazzini, L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes 2023, 14, 1658. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.; Andrews, S.J.; Tripathy, S.J. Shared Genetic Risk Loci between Alzheimer’s Disease and Related Dementias, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Alzheimer’s Res. Ther. 2023, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome Dysfunction as a Cause of Neurodegenerative Diseases: Lessons from Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J. CGAS–STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in Neurological Disorders and Emerging Complement-Targeted Therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, I.; Fluiter, K.; Boziki, M.; Grigoriadis, N.; Baas, F. Complement in Nervous System Disease. Front. Cell. Neurosci. 2023, 17, 1268023. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial Phagocytosis of Neurons in Neurodegeneration, and Its Regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Cao, W.; Cao, Z.; Tian, Y.; Zhang, L.; Wang, W.; Tang, L.; Xu, C.; Fan, D. Neutrophils Are Associated with Higher Risk of Incident Amyotrophic Lateral Sclerosis in a BMI-and Age-dependent Manner. Ann. Neurol. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Tondo, G.; Aprile, D.; De Marchi, F.; Sarasso, B.; Serra, P.; Borasio, G.; Rojo, E.; Arenillas, J.F.; Comi, C. Investigating the Prognostic Role of Peripheral Inflammatory Markers in Mild Cognitive Impairment. J. Clin. Med. 2023, 12, 4298. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.A.; Vilensky, J.A. Encephalitis Lethargica: 100 Years after the Epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Alfahad, T.; Nath, A. Retroviruses and Amyotrophic Lateral Sclerosis. Antiviral Res. 2013, 99, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of Active Loci of a Human Endogenous Retrovirus in Neurons of Patients with Amyotrophic Lateral Sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Puma, D.D.L.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Z. COVID-19 and the Risk of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2022, 9, 1953–1961. [Google Scholar] [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, P.N.; Heikkilä, N.; Lindbohm, J.V.; Hakulinen, C.; Vahtera, J.; Elovainio, M.; Suominen, S.; Väänänen, A.; Koskinen, A.; Nyberg, S.T. Hospital-Treated Infectious Diseases and the Risk of Dementia: A Large, Multicohort, Observational Study with a Replication Cohort. Lancet Infect. Dis. 2021, 21, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Y.; Wang, Z.; Xie, G.; Liu, M.; Yuan, B.; Chai, H.; Wang, W.; Cheng, P. Implications of Gut Microbiota in Neurodegenerative Diseases. Front. Immunol. 2022, 13, 785644. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, D.; Bozzi Cionci, N.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggìa, F.; Lucenti, M.A.; Bersano, E.; Cantello, R. A Prospective Longitudinal Study on the Microbiota Composition in Amyotrophic Lateral Sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef]
- Papić, E.; Rački, V.; Hero, M.; Tomić, Z.; Starčević-Čižmarević, N.; Kovanda, A.; Kapović, M.; Hauser, G.; Peterlin, B.; Vuletić, V. The Effects of Microbiota Abundance on Symptom Severity in Parkinson’s Disease: A Systematic Review. Front. Aging Neurosci. 2022, 14, 1020172. [Google Scholar] [CrossRef]
- Fülling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus…. Neuron 2019, 101, 998–1002. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Fung, T.C.; Vuong, H.E.; Luna, C.D.G.; Pronovost, G.N.; Aleksandrova, A.A.; Riley, N.G.; Vavilina, A.; McGinn, J.; Rendon, T.; Forrest, L.R. Intestinal Serotonin and Fluoxetine Exposure Modulate Bacterial Colonization in the Gut. Nat. Microbiol. 2019, 4, 2064–2073. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine Pathway Metabolism and the Microbiota-Gut-Brain Axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Chu, H.; Mazmanian, S.K. Innate Immune Recognition of the Microbiota Promotes Host-Microbial Symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.-Y.; Pietilainen, O. C9orf72 Suppresses Systemic and Neural Inflammation Induced by Gut Bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H. Role of TLR4 in the Gut-Brain Axis in Parkinson’s Disease: A Translational Study from Men to Mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N. Interplay between Immunity and Amyotrophic Lateral Sclerosis: Clinical Impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A. Genome-Wide Association Study Identifies Variants at CLU and PICALM Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. Alzheimer Genetic Analysis G (2013) TREM2 Variants in Alzheimer’s Disease. N Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, K. TREM2 and Neurodegenerative Disease. N Engl. J. Med. 2013, 369, 1568–1569. [Google Scholar]
- Huang, K.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J. A Common Haplotype Lowers PU. 1 Expression in Myeloid Cells and Delays Onset of Alzheimer’s Disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef]
- Thambisetty, M.; Beason-Held, L.L.; An, Y.; Kraut, M.; Nalls, M.; Hernandez, D.G.; Singleton, A.B.; Zonderman, A.B.; Ferrucci, L.; Lovestone, S.; et al. Alzheimer Risk Variant CLU and Brain Function during Aging. Biol. Psychiatry 2013, 73, 399–405. [Google Scholar] [CrossRef]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 Mutations Implicated in Neurodegeneration Impair Cell Surface Transport and Phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Gerrits, E.; Brouwer, N.; Kooistra, S.M.; Woodbury, M.E.; Vermeiren, Y.; Lambourne, M.; Mulder, J.; Kummer, M.; Möller, T.; Biber, K.; et al. Distinct Amyloid-β and Tau-Associated Microglia Profiles in Alzheimer’s Disease. Acta Neuropathol. 2021, 141, 681–696. [Google Scholar] [CrossRef]
- Solito, E.; Sastre, M. Microglia Function in Alzheimer’s Disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Melief, J.; Kooijman, L.; Huitinga, I.; Klooster, J.; Bossers, K.; Hol, E.M. Acute Isolation and Transcriptome Characterization of Cortical Astrocytes and Microglia from Young and Aged Mice. Neurobiol. Aging 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Zlokovic, B. V The Role of the Cell Surface LRP and Soluble LRP in Blood-Brain Barrier Aβ Clearance in Alzheimer’s Disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C. Human ApoE Isoforms Differentially Regulate Brain Amyloid-Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’loughlin, E.; Xu, Y.; Fanek, Z. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N. Loss of TREM2 Function Increases Amyloid Seeding but Reduces Plaque-Associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE Mediated Neuroinflammation and Neurodegeneration in Alzheimer’s Disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef] [PubMed]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young Microglia Restore Amyloid Plaque Clearance of Aged Microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The Antimicrobial Protection Hypothesis of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019: A Review. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Rai, S.N.; Tiwari, N.; Singh, P.; Singh, A.K.; Mishra, D.; Imran, M.; Singh, S.; Hooshmandi, E.; Vamanu, E.; Singh, S.K.; et al. Exploring the Paradox of COVID-19 in Neurological Complications with Emphasis on Parkinson’s and Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 3012778. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Karten, B.; Peake, K.B.; Vance, J.E. Mechanisms and Consequences of Impaired Lipid Trafficking in Niemann–Pick Type C1-Deficient Mammalian Cells. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Wu, X.; Du, X.; Yao, X.; Zhao, X.; Lee, J.; Yang, H.; Yan, N. Structural Basis of Low-PH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 2020, 182, 98–111. [Google Scholar] [CrossRef]
- Loftus, S.K.; Erickson, R.P.; Walkley, S.U.; Bryant, M.A.; Incao, A.; Heidenreich, R.A.; Pavan, W.J. Rescue of Neurodegeneration in Niemann–Pick C Mice by a Prion-Promoter-Driven Npc1 CDNA Transgene. Hum. Mol. Genet. 2002, 11, 3107–3114. [Google Scholar] [CrossRef]
- Chen, G.; Li, H.; Chen, Y.; Gu, X.; Duan, S. Decreased Estradiol Release from Astrocytes Contributes to the Neurodegeneration in a Mouse Model of Niemann-Pick Disease Type C. Glia 2007, 55, 1509–1518. [Google Scholar] [CrossRef]
- Lopez, M.E.; Klein, A.D.; Dimbil, U.J.; Scott, M.P. Anatomically Defined Neuron-Based Rescue of Neurodegenerative Niemann–Pick Type C Disorder. J. Neurosci. 2011, 31, 4367–4378. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.A.; Watkins-Chow, D.E.; Palladino, G.; Deutsch, G.; Chandran, K.; Pavan, W.J.; Erickson, R.P. In Niemann-Pick C1 Mouse Models, Glial-Only Expression of the Normal Gene Extends Survival Much Further than Do Changes in Genetic Background or Treatment with Hydroxypropyl-Beta-Cyclodextrin. Gene 2018, 643, 117–123. [Google Scholar] [CrossRef]
- Zhang, Q.-G.; Wang, R.; Khan, M.; Mahesh, V.; Brann, D.W. Role of Dickkopf-1, an Antagonist of the Wnt/β-Catenin Signaling Pathway, in Estrogen-Induced Neuroprotection and Attenuation of Tau Phosphorylation. J. Neurosci. 2008, 28, 8430–8441. [Google Scholar] [CrossRef]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P. Loss of NPC1 Enhances Phagocytic Uptake and Impairs Lipid Trafficking in Microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef]
- Heiden, D.L.; Monogue, B.; Ali, M.D.H.; Beckham, J.D. A Functional Role for Alpha-Synuclein in Neuroimmune Responses. J. Neuroimmunol. 2023, 376, 578047. [Google Scholar] [CrossRef]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int. J. Mol. Sci. 2023, 24, 2477. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.H.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.C.; Rudd, J.A. Intra-gastrointestinal Amyloid-β1–42 Oligomers Perturb Enteric Function and Induce Alzheimer’s Disease Pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum Promotes Drosophila Systemic Growth by Modulating Hormonal Signals through TOR-Dependent Nutrient Sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef]
- Wu, G.; Jiang, Z.; Pu, Y.; Chen, S.; Wang, T.; Wang, Y.; Xu, X.; Wang, S.; Jin, M.; Yao, Y. Serum Short-Chain Fatty Acids and Its Correlation with Motor and Non-Motor Symptoms in Parkinson’s Disease Patients. BMC Neurol. 2022, 22, 13. [Google Scholar] [CrossRef]
- Murros, K.E.; Huynh, V.A.; Takala, T.M.; Saris, P.E.J. Desulfovibrio Bacteria Are Associated with Parkinson’s Disease. Front. Cell. Infect. Microbiol. 2021, 11, 652617. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey III, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. FASEB J. 2015, 29, 1395. [Google Scholar] [CrossRef]
- Clarke, M.T.M.; Brinkmalm, A.; Foiani, M.S.; Woollacott, I.O.C.; Heller, C.; Heslegrave, A.; Keshavan, A.; Fox, N.C.; Schott, J.M.; Warren, J.D.; et al. CSF Synaptic Protein Concentrations Are Raised in Those with Atypical Alzheimer’s Disease but Not Frontotemporal Dementia. Alzheimer’s Res. Ther. 2019, 11, 105. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y. Altered Gut Microbiota and Inflammatory Cytokine Responses in Patients with Parkinson’s Disease. J. Neuroinflammation 2019, 16, 129. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. The Role of Environmental Exposures in Neurodegeneration and Neurodegenerative Diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, A.G.; Rhodes, S.L.; Cockburn, M.; Ritz, B.; Bronstein, J.M. Aldehyde Dehydrogenase Variation Enhances Effect of Pesticides Associated with Parkinson Disease. Neurology 2014, 82, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O’Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A. Aldehyde Dehydrogenase Inhibition as a Pathogenic Mechanism in Parkinson Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Gatto, N.M.; Cockburn, M.; Bronstein, J.; Manthripragada, A.D.; Ritz, B. Well-Water Consumption and Parkinson’s Disease in Rural California. Environ. Health Perspect. 2009, 117, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Tarbutton, G.L.; Levin, J.L.; Plotkin, G.M.; Lowry, L.K.; Nalbone, J.T.; Shepherd, S. Pesticide/Environmental Exposures and Parkinson’s Disease in East Texas. J. Agromedicine 2008, 13, 37–48. [Google Scholar] [CrossRef]
- Manthripragada, A.D.; Costello, S.; Cockburn, M.G.; Bronstein, J.M.; Ritz, B. Paraoxonase 1 (PON1), Agricultural Organophosphate Exposure, and Parkinson Disease. Epidemiology 2010, 21, 87. [Google Scholar] [CrossRef]
- Porras, G.; Ruiz, S.; Maestro, I.; Borrego-Hernández, D.; Redondo, A.G.; Martínez, A.; Martín-Requero, Á. Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts. Int. J. Mol. Sci. 2023, 24, 2847. [Google Scholar] [CrossRef]
- Voorhees, J.R.; Remy, M.T.; Erickson, C.M.; Dutca, L.M.; Brat, D.J.; Pieper, A.A. Occupational-like Organophosphate Exposure Disrupts Microglia and Accelerates Deficits in a Rat Model of Alzheimer’s Disease. NPJ Aging Mech. Dis. 2019, 5, 3. [Google Scholar] [CrossRef]
- Andrew, A.; Zhou, J.; Gui, J.; Harrison, A.; Shi, X.; Li, M.; Guetti, B.; Nathan, R.; Tischbein, M.; Pioro, E.P. Pesticides Applied to Crops and Amyotrophic Lateral Sclerosis Risk in the US. Neurotoxicology 2021, 87, 128–135. [Google Scholar] [CrossRef]
- Landers, J.E.; Shi, L.; Cho, T.-J.; Glass, J.D.; Shaw, C.E.; Leigh, P.N.; Diekstra, F.; Polak, M.; Rodriguez-Leyva, I.; Niemann, S. A Common Haplotype within the PON1 Promoter Region Is Associated with Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 306–314. [Google Scholar] [CrossRef]
- Arnold, G.E.; Manchester, J.I.; Townsend, B.D.; Ornstein, R.L. Investigation of Domain Motions in Bacteriophage T4 Lysozyme. J. Biomol. Struct. Dyn. 1994, 12, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in Amyotrophic Lateral Sclerosis: Multifunctional Adaptor Protein at the Crossroads of Different Neuroprotective Mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Lill, C.M.; Roehr, J.T.; McQueen, M.B.; Kavvoura, F.K.; Bagade, S.; Schjeide, B.-M.M.; Schjeide, L.M.; Meissner, E.; Zauft, U.; Allen, N.C. Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson’s Disease Genetics: The PDGene Database. PLoS Genet. 2012, 8, e1002548. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.M.; Kirkley, K.S.; Chatterjee, D.; Aboellail, T.A.; Smeyne, R.J.; Tjalkens, R.B. Microglia-specific Knock-out of NF-κB/IKK2 Increases the Accumulation of Misfolded A-synuclein through the Inhibition of P62/Sequestosome-1-dependent Autophagy in the Rotenone Model of Parkinson’s Disease. Glia 2023, 71, 2154–2179. [Google Scholar] [CrossRef]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune Connections in Aging and Neurodegenerative Diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein Aggregates in the Pathogenesis, Prognosis, and Therapeutics for Neurodegenerative Diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef]
- Arevalo-Villalobos, J.I.; Rosales-Mendoza, S.; Zarazua, S. Immunotherapies for Neurodegenerative Diseases: Current Status and Potential of Plant-Made Biopharmaceuticals. Expert Rev. Vaccines 2017, 16, 151–159. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, J.; Li, X.; Yuan, L.; Pan, Y.; Chen, X.; Gao, T.; Qiao, J.; Qi, J. A Novel Antibody Targeting Sequence 31–35 in Amyloid β Protein Attenuates Alzheimer’s Disease-related Neuronal Damage. Hippocampus 2017, 27, 122–133. [Google Scholar] [CrossRef]
- Minami, S.S.; Sidahmed, E.; Aid, S.; Shimoji, M.; Niikura, T.; Mocchetti, I.; Rebeck, G.W.; Prendergast, J.S.; Dealwis, C.; Wetzel, R. Therapeutic versus Neuroinflammatory Effects of Passive Immunization Is Dependent on Aβ/Amyloid Burden in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neuroinflammation 2010, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; Von Hehn, C. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in Early Alzheimer’s Disease. New Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C. The Clinical Dementia Rating (CDR): Current Version and Scoring Rules. Neurology 1993, 43, 2412–2414. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.J.; Bullock, R.; Jones, R.W.; Wilkinson, D.; Paterson, K.R.; Jenkins, L.; Millais, S.B.; Donoghue, S. Evaluation of the Safety and Immunogenicity of Synthetic Aβ42 (AN1792) in Patients with AD. Neurology 2005, 64, 94–101. [Google Scholar] [CrossRef]
- Paquet, C.; Nicoll, J.A.R.; Love, S.; Mouton-Liger, F.; Holmes, C.; Hugon, J.; Boche, D. Downregulated Apoptosis and Autophagy after Anti-Aβ Immunotherapy in Alzheimer’s Disease. Brain Pathol. 2018, 28, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent Neuropathological Effects 14 Years Following Amyloid-β Immunization in Alzheimer’s Disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Sadowsky, C.; Le Prince Leterme, G.; Booth, K.; Peng, Y.; Marek, K.; Ketter, N.; Liu, E.; Wyman, B.T.; Jackson, N. Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Individuals with Early Alzheimer’s Disease: Amyloid Imaging Positron Emission Tomography and Safety Results from a Phase 2 Study. J. Prev. Alzheimer’s Dis. 2016, 3, 75–84. [Google Scholar] [CrossRef]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and Sequencing of the CDNA Encoding a Core Protein of the Paired Helical Filament of Alzheimer Disease: Identification as the Microtubule-Associated Protein Tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J. Tau and Aβ Imaging, CSF Measures, and Cognition in Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 338ra66. [Google Scholar] [CrossRef]
- Katsinelos, T.; Tuck, B.J.; Mukadam, A.S.; McEwan, W.A. The Role of Antibodies and Their Receptors in Protection against Ordered Protein Assembly in Neurodegeneration. Front. Immunol. 2019, 10, 1139. [Google Scholar] [CrossRef]
- Novak, P.; Kovacech, B.; Katina, S.; Schmidt, R.; Scheltens, P.; Kontsekova, E.; Ropele, S.; Fialova, L.; Kramberger, M.; Paulenka-Ivanovova, N. ADAMANT: A Placebo-Controlled Randomized Phase 2 Study of AADvac1, an Active Immunotherapy against Pathological Tau in Alzheimer’s Disease. Nat. Aging 2021, 1, 521–534. [Google Scholar] [CrossRef]
- Huang, Y.; Xie, X.; Ji, M.; Yu, X.; Zhu, J.; Zhang, L.; Liu, X.; Wei, C.; Li, G.; Liu, R. Naturally Occurring Autoantibodies against α-Synuclein Rescues Memory and Motor Deficits and Attenuates α-Synuclein Pathology in Mouse Model of Parkinson’s Disease. Neurobiol. Dis. 2019, 124, 202–217. [Google Scholar] [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. New Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T. Randomized Phase I Clinical Trial of Anti–A-synuclein Antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.-H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Investigators, H.S.G.R. Safety, Tolerability, and Efficacy of PBT2 in Huntington’s Disease: A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2015, 14, 39–47. [Google Scholar]
- Poulin-Brière, A.; Rezaei, E.; Pozzi, S. Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front. Neurosci. 2021, 15, 790114. [Google Scholar] [CrossRef]
- Milligan, C.; Atassi, N.; Babu, S.; Barohn, R.J.; Caress, J.B.; Cudkowicz, M.E.; Evora, A.; Hawkins, G.A.; Wosiski-Kuhn, M.; Macklin, E.A. Tocilizumab Is Safe and Tolerable and Reduces C-reactive Protein Concentrations in the Plasma and Cerebrospinal Fluid of ALS Patients. Muscle Nerve 2021, 64, 309–320. [Google Scholar] [CrossRef]
- Meininger, V.; Genge, A.; van den Berg, L.H.; Robberecht, W.; Ludolph, A.; Chio, A.; Kim, S.H.; Leigh, P.N.; Kiernan, M.C.; Shefner, J.M. Safety and Efficacy of Ozanezumab in Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2017, 16, 208–216. [Google Scholar] [CrossRef]
- Group, A.-F.R. Follow-up Evaluation of Cognitive Function in the Randomized Alzheimer’s Disease Anti-Inflammatory Prevention Trial and Its Follow-up Study. Alzheimer’s Dement. 2015, 11, 216–225. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R. Etanercept in Alzheimer Disease: A Randomized, Placebo-Controlled, Double-Blind, Phase 2 Trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef]
- Alves, S.; Churlaud, G.; Audrain, M.; Michaelsen-Preusse, K.; Fol, R.; Souchet, B.; Braudeau, J.; Korte, M.; Klatzmann, D.; Cartier, N. Interleukin-2 Improves Amyloid Pathology, Synaptic Failure and Memory in Alzheimer’s Disease Mice. Brain 2017, 140, 826–842. [Google Scholar] [CrossRef]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Yokoyama, I.; Mosley, R.L.; Gendelman, H.E. Granulocyte-Macrophage Colony-Stimulating Factor Neuroprotective Activities in Alzheimer’s Disease Mice. J. Neuroimmunol. 2018, 319, 80–92. [Google Scholar] [CrossRef]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O’Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D. Safety and Efficacy of Sargramostim (GM-CSF) in the Treatment of Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12158. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G. Evaluation of the Safety and Immunomodulatory Effects of Sargramostim in a Randomized, Double-Blind Phase 1 Clinical Parkinson’s Disease Trial. NPJ Park. Dis. 2017, 3, 10. [Google Scholar]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; De Biasi, S.; Banchelli, F.; Martinelli, I.; Simonini, C.; Lo Tartaro, D.; Vicini, R.; Fini, N. Randomized, Double-Blind, Placebo-Controlled Trial of Rapamycin in Amyotrophic Lateral Sclerosis. Nat. Commun. 2023, 14, 4970. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Berry, J.D.; Macklin, E.A.; Beers, D.R.; Mendoza, P.A.; Zhao, W.; Thome, A.D.; Triolo, F.; Moon, J.J.; Paganoni, S.; et al. Combined Regulatory T-Lymphocyte and IL-2 Treatment Is Safe, Tolerable, and Biologically Active for 1 Year in Persons with Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2022, 9. [Google Scholar] [CrossRef]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef]
- Lee, J.D.; Kumar, V.; Fung, J.N.T.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological Inhibition of Complement C5a-C5a1 Receptor Signalling Ameliorates Disease Pathology in the HSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef]
- De Marchi, F.; Mareschi, K.; Ferrero, I.; Cantello, R.; Fagioli, F.; Mazzini, L. Effect of Mesenchymal Stromal Cell Transplantation on Long-Term Survival in Amyotrophic Lateral Sclerosis. Cytotherapy 2023, 25, 798–802. [Google Scholar] [CrossRef]
- Francesca, S.; De Marchi, F.; Mazzini, L.; Bendotti, C. Cell Therapy in ALS: An Update on Preclinical and Clinical Studies. Brain Res. Bull. 2023, 194, 64–81. [Google Scholar]
- Brody, M.; Agronin, M.; Herskowitz, B.J.; Bookheimer, S.Y.; Small, G.W.; Hitchinson, B.; Ramdas, K.; Wishard, T.; McInerney, K.F.; Vellas, B. Results and Insights from a Phase I Clinical Trial of Lomecel-B for Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 261–273. [Google Scholar] [CrossRef]
- Cai, J.; Wu, J.; Wang, J.; Li, Y.; Hu, X.; Luo, S.; Xiang, D. Extracellular Vesicles Derived from Different Sources of Mesenchymal Stem Cells: Therapeutic Effects and Translational Potential. Cell Biosci. 2020, 10, 69. [Google Scholar] [CrossRef]
- Qian, X.; An, N.; Ren, Y.; Yang, C.; Zhang, X.; Li, L. Immunosuppressive Effects of Mesenchymal Stem Cells-Derived Exosomes. Stem Cell Rev. Rep. 2021, 17, 411–427. [Google Scholar] [CrossRef]
- Pu, Y.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Hashimoto, K. Antibiotic-Induced Microbiome Depletion Protects against MPTP-Induced Dopaminergic Neurotoxicity in the Brain. Aging 2019, 11, 6915. [Google Scholar] [CrossRef]
- Li, W.; Lesuisse, C.; Xu, Y.; Troncoso, J.C.; Price, D.L.; Lee, M.K. Stabilization of α-Synuclein Protein with Aging and Familial Parkinson’s Disease-Linked A53T Mutation. J. Neurosci. 2004, 24, 7400–7409. [Google Scholar] [CrossRef]
- Ojha, S.; Patil, N.; Jain, M.; Kole, C.; Kaushik, P. Probiotics for Neurodegenerative Diseases: A Systemic Review. Microorganisms 2023, 11, 1083. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Gu, Y.; Mejia-Santana, H.; Cote, L.; Marder, K.S.; Scarmeas, N. The Association between Mediterranean Diet Adherence and Parkinson’s Disease. Mov. Disord. 2012, 27, 771–774. [Google Scholar] [CrossRef]
- Mandrioli, J.; Amedei, A.; Cammarota, G.; Niccolai, E.; Zucchi, E.; D’Amico, R.; Ricci, F.; Quaranta, G.; Spanu, T.; Masucci, L. FETR-ALS Study Protocol: A Randomized Clinical Trial of Fecal Microbiota Transplantation in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 1021. [Google Scholar] [CrossRef]
- Mohovic, N.; Peradinovic, J.; Markovinovic, A.; Cimbro, R.; Minic, Z.; Dominovic, M.; Jakovac, P.; Nimac, J.; Rogelj, B.; Munitic, P. Neuroimmune characterization of optineurin insufficiency mouse model during ageing. Sci. Rep. 2023, 13, 11840. [Google Scholar] [CrossRef]
- Xue, L.-J.; Yang, X.-Z.; Tong, Q.; Shen, P.; Ma, S.-J.; Wu, S.-N.; Zheng, J.-L.; Wang, H.-G. Fecal Microbiota Transplantation Therapy for Parkinson’s Disease: A Preliminary Study. Medicine 2020, 99, PMC7458210. [Google Scholar] [CrossRef]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J. Genome Sequencing Analysis Identifies New Loci Associated with Lewy Body Dementia and Provides Insights into Its Genetic Architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Bukhbinder, A.S.; Ling, Y.; Hasan, O.; Jiang, X.; Kim, Y.; Phelps, K.N.; Schmandt, R.E.; Amran, A.; Coburn, R.; Ramesh, S. Risk of Alzheimer’s Disease Following Influenza Vaccination: A Claims-Based Cohort Study Using Propensity Score Matching. J. Alzheimer’s Dis. 2022, 88, 1061–1074. [Google Scholar] [CrossRef]
- Maximova, A.; Werry, E.L.; Kassiou, M. Senolytics: A Novel Strategy for Neuroprotection in ALS? Int. J. Mol. Sci. 2021, 22, 12078. [Google Scholar] [CrossRef]
- Miller, S.J.; Campbell, C.E.; Jimenez-Corea, H.A.; Wu, G.-H.; Logan, R. Neuroglial Senescence, α-Synucleinopathy, and the Therapeutic Potential of Senolytics in Parkinson’s Disease. Front. Neurosci. 2022, 16, 824191. [Google Scholar] [CrossRef]
- Ahmadi Rastegar, D.; Hughes, L.P.; Perera, G.; Keshiya, S.; Zhong, S.; Gao, J.; Halliday, G.M.; Schüle, B.; Dzamko, N. Effect of LRRK2 Protein and Activity on Stimulated Cytokines in Human Monocytes and Macrophages. NPJ Park. Dis. 2022, 8, 34. [Google Scholar] [CrossRef]
- Russo, I.; Kaganovich, A.; Ding, J.; Landeck, N.; Mamais, A.; Varanita, T.; Biosa, A.; Tessari, I.; Bubacco, L.; Greggio, E. Transcriptome Analysis of LRRK2 Knock-out Microglia Cells Reveals Alterations of Inflammatory-and Oxidative Stress-Related Pathways upon Treatment with α-Synuclein Fibrils. Neurobiol. Dis. 2019, 129, 67–78. [Google Scholar] [CrossRef]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-Rich Repeat Kinase 2 Positively Regulates Inflammation and down-Regulates NF-ΚB P50 Signaling in Cultured Microglia Cells. J. Neuroinflammation 2015, 12, 230. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Mutti, V.; Carini, G.; Filippini, A.; Castrezzati, S.; Giugno, L.; Gennarelli, M.; Russo, I. LRRK2 Kinase Inhibition Attenuates Neuroinflammation and Cytotoxicity in Animal Models of Alzheimer’s and Parkinson’s Disease-Related Neuroinflammation. Cells 2023, 12, 1799. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V. V Preclinical and Clinical Evaluation of the LRRK2 Inhibitor DNL201 for Parkinson’s Disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; O’Donnell, P.; Drobatz, K.; Gurda, B. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J. Pharmacol. Exp. Ther. 2016, 358, 254–261. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease Ameliorates Neuronal Cholesterol and Glycosphingolipid Storage and Disease Progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A. Intrathecal 2-Hydroxypropyl-β-Cyclodextrin Decreases Neurological Disease Progression in Niemann-Pick Disease, Type C1: A Non-Randomised, Open-Label, Phase 1–2 Trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- Las Heras, M.; Szenfeld, B.; Ballout, R.A.; Buratti, E.; Zanlungo, S.; Dardis, A.; Klein, A.D. Understanding the Phenotypic Variability in Niemann-Pick Disease Type C (NPC): A Need for Precision Medicine. NPJ Genom. Med. 2023, 8, 21. [Google Scholar] [CrossRef]
- Santos-Lozano, A.; García, D.V.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Gadea, G.N.; Lucia, A. Niemann-Pick Disease Treatment: A Systematic Review of Clinical Trials. Ann. Transl. Med. 2015, 3, 360. [Google Scholar] [PubMed]
- Szego, E.M.; Malz, L.; Bernhardt, N.; Rösen-Wolff, A.; Falkenburger, B.H.; Luksch, H. Constitutively Active STING Causes Neuroinflammation and Degeneration of Dopaminergic Neurons in Mice. Elife 2022, 11, e81943. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Phaneuf, D.; Julien, J.-P. Withaferin-A Treatment Alleviates TAR DNA-Binding Protein-43 Pathology and Improves Cognitive Function in a Mouse Model of FTLD. Neurotherapeutics 2021, 18, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Malhotra, S.; Shinohara, M.L.; Montalban, X.; Comabella, M. Targeting Inflammasomes to Treat Neurological Diseases. Ann. Neurol. 2021, 90, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-Interacting Protein Kinase 1 (RIPK1) as a Therapeutic Target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L. RIPK1 Mediates Axonal Degeneration by Promoting Inflammation and Necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the Role of the NLRP3 Inflammasome in Multiple Sclerosis: Pathogenesis, Diagnosis, and Therapeutics. Front. Mol. Neurosci. 2022, 15, 894298. [Google Scholar]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.-P.; Dupré, N. From Animal Models to Human Disease: A Genetic Approach for Personalized Medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- Mckean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef]
- Mohovic, N.; Peradinovic, J.; Markovinovic, A.; Cimbro, R.; Minic, Z.; Dominovic, M.; Jakovac, H.; Nimac, J.; Rogelj, B.; Munitic, I. Neuroimmune Characterization of Optineurin Insufficiency Mouse Model during Ageing. bioRxiv 2023. bioRxiv:2023-03. [Google Scholar] [CrossRef]
- Brenner, D.; Sieverding, K.; Bruno, C.; Lüningschrör, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhäuser, C.; et al. Heterozygous Tbk1 Loss Has Opposing Effects in Early and Late Stages of ALS in Mice. J. Exp. Med. 2019, 216, 267–278. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. https://doi.org/10.3390/biomedicines11102793
De Marchi F, Munitic I, Vidatic L, Papić E, Rački V, Nimac J, Jurak I, Novotni G, Rogelj B, Vuletic V, et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines. 2023; 11(10):2793. https://doi.org/10.3390/biomedicines11102793
Chicago/Turabian StyleDe Marchi, Fabiola, Ivana Munitic, Lea Vidatic, Eliša Papić, Valentino Rački, Jerneja Nimac, Igor Jurak, Gabriela Novotni, Boris Rogelj, Vladimira Vuletic, and et al. 2023. "Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders" Biomedicines 11, no. 10: 2793. https://doi.org/10.3390/biomedicines11102793
APA StyleDe Marchi, F., Munitic, I., Vidatic, L., Papić, E., Rački, V., Nimac, J., Jurak, I., Novotni, G., Rogelj, B., Vuletic, V., Liscic, R. M., Cannon, J. R., Buratti, E., Mazzini, L., & Hecimovic, S. (2023). Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines, 11(10), 2793. https://doi.org/10.3390/biomedicines11102793