
Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Line Culture
2.2. Animals
2.3. Drugs
2.4. Bone Marrow-Derived Macrophages
2.5. Bone Marrow-Derived MDSCs
2.6. Flow Cytometry Analysis of Immune Cells
2.7. In Vivo Tumor Models
2.8. Antigen-Specific T-Cell Response Detection by IFN-γ ELISpot
2.9. Quantitative PCR
2.10. Statistical Analysis
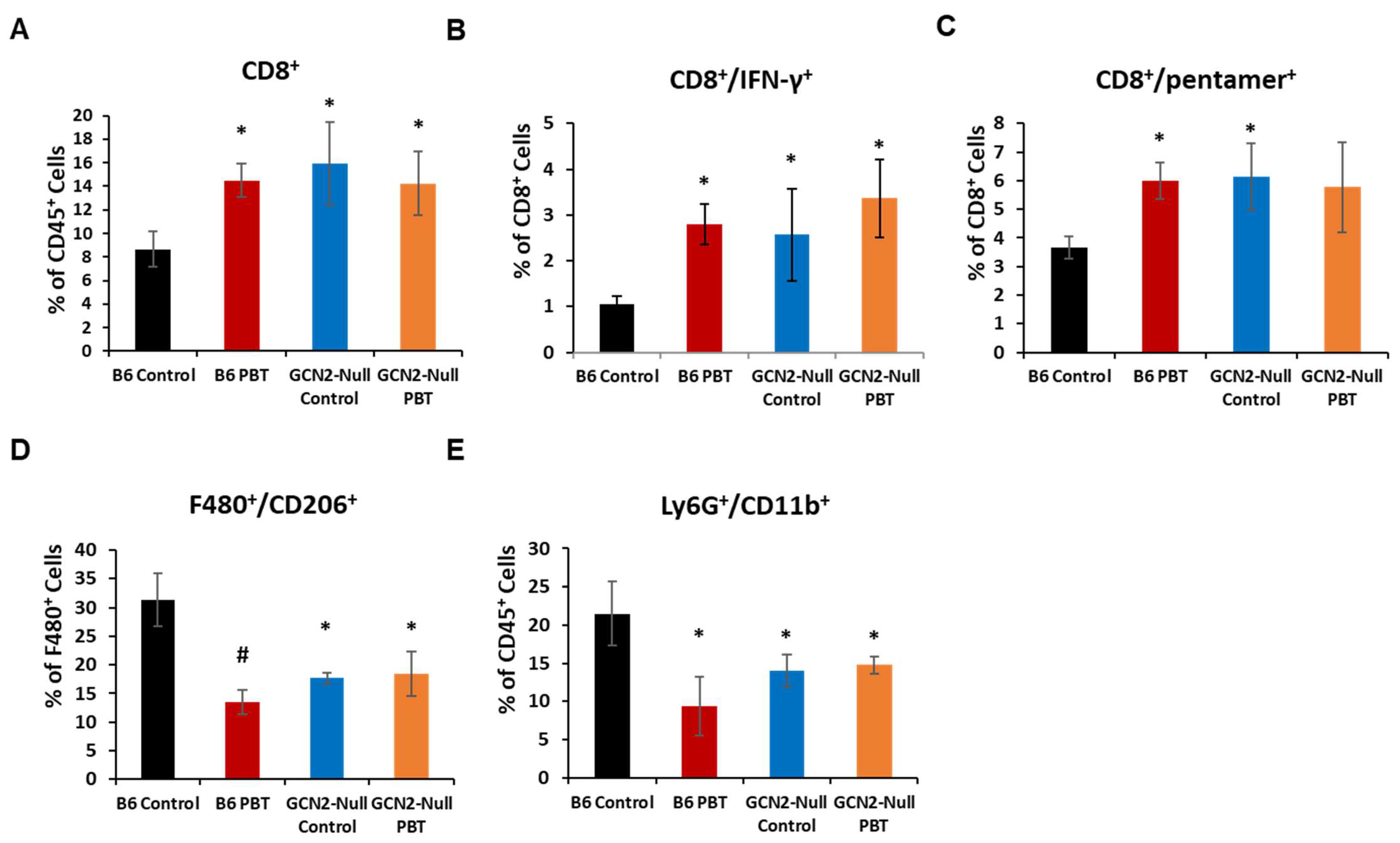
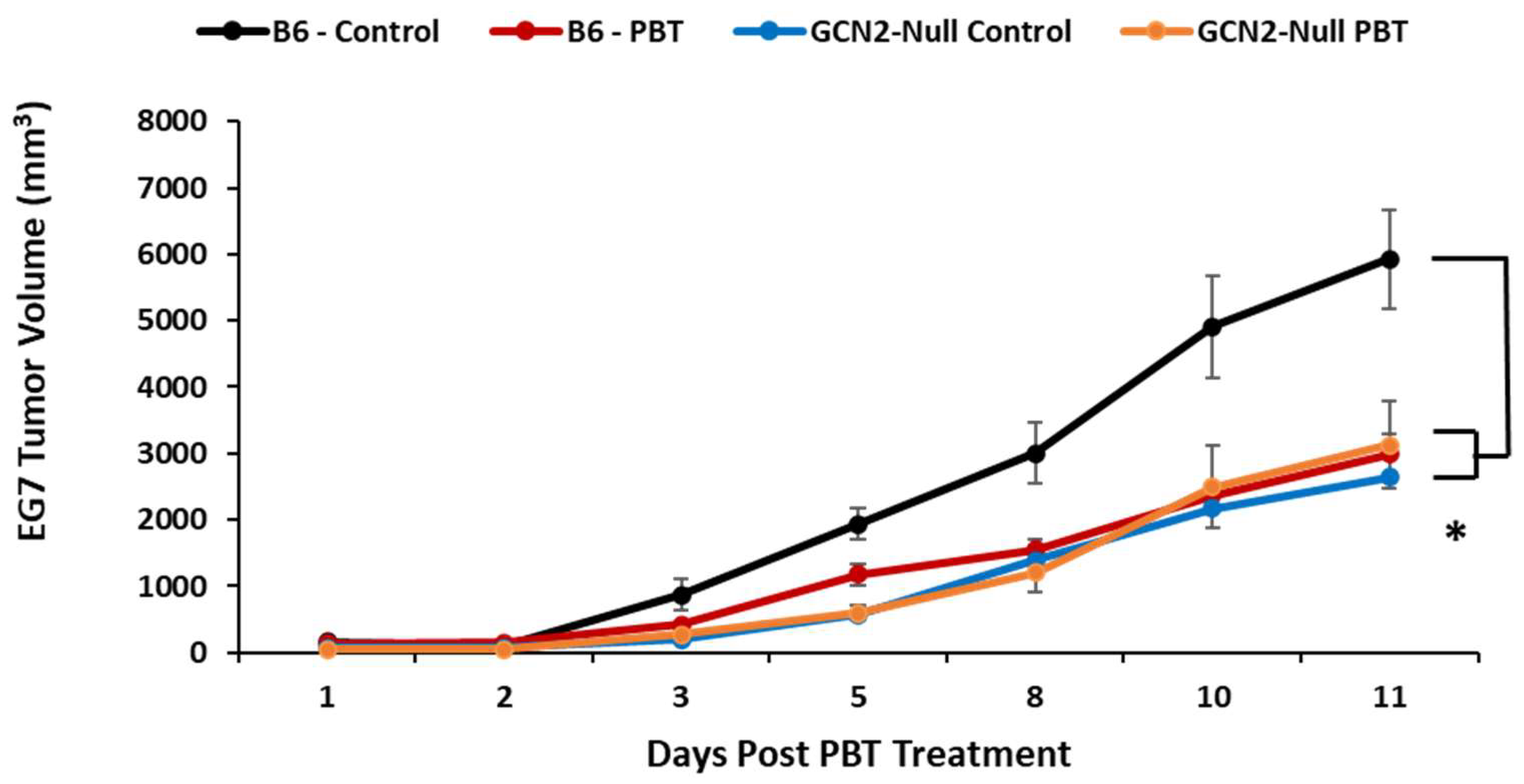
3. Results
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walls, J.; Sinclair, L.; Finlay, D. Nutrient sensing, signal transduction and immune responses. Semin. Immunol. 2016, 28, 396–407. [Google Scholar] [CrossRef]
- Qiu, H.; Dong, J.; Hu, C.; Francklyn, C.S.; Hinnebusch, A.G. The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J. 2001, 20, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, R.J.; Hinnebusch, A.G. Translation of the yeast transcriptional activator GCN4 is stimulated by purine limitation: Implications for activation of the protein kinase GCN2. Mol. Cell. Biol. 1993, 13, 5099–5111. [Google Scholar] [PubMed]
- Gold, L.T.; Masson, G.R. GCN2: Roles in tumour development and progression. Biochem. Soc. Trans. 2022, 50, 737–745. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salinas, C.S.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Adomavicius, T.; Guaita, M.; Zhou, Y.; Jennings, M.D.; Latif, Z.; Roseman, A.M.; Pavitt, G.D. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 2019, 10, 2136. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Gay, D.; Uthe, F.W.; Denk, S.; Paauwe, M.; Matthes, N.; Diefenbacher, M.E.; Bryson, S.; Warrander, F.C.; Erhard, F.; et al. A MYC-GCN2-eIF2α negative feedback loop limits protein synthesis to prevent MYC-dependent apoptosis in colorectal cancer. Nat. Cell Biol. 2019, 21, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Chen, M.S.; Chou, Y.C.; Ueng, Y.F.; Yin, P.H.; Yeh, T.S.; Lee, H.C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132–74151. [Google Scholar] [CrossRef]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Halaby, M.J.; Hezaveh, K.; Lamorte, S.; Ciudad, M.T.; Kloetgen, A.; MacLeod, B.L.; Guo, M.; Chakravarthy, A.; Medina, T.D.S.; Ugel, S.; et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci. Immunol. 2019, 4, eaax8189. [Google Scholar] [CrossRef]
- Zhao, C.; Guo, H.; Hou, Y.; Lei, T.; Wei, D.; Zhao, Y. Multiple Roles of the Stress Sensor GCN2 in Immune Cells. Int. J. Mol. Sci. 2023, 24, 4285. [Google Scholar] [CrossRef]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Cervelli, M.; Pietropaoli, S.; Signore, F.; Amendola, R.; Mariottini, P. Polyamines metabolism and breast cancer: State of the art and perspectives. Breast Cancer Res. Treat. 2014, 148, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Minton, A.; Peters, M.C.; Phanstiel, O., IV; Gilmour, S.K. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget 2017, 8, 84140–84152. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Holbert, C.E.; Cullen, M.T.; Casero, R.A., Jr.; Stewart, T.M. Polyamines in cancer: Integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 2022, 22, 467–480. [Google Scholar] [CrossRef]
- Geck, R.C.; Foley, J.R.; Murray Stewart, T.M.; Asara, J.M.; Casero, R.A., Jr.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Biol. Chem. 2020, 295, 6263–6277. [Google Scholar] [CrossRef]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef]
- Forshell, T.P.; Rimpi, S.; Nilsson, J.A. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. 2010, 3, 140–147. [Google Scholar] [CrossRef]
- Bachrach, U.; Seiler, N. Formation of acetylpolyamines and putrescine from spermidine by normal and transformed chick embryo fibroblasts. Cancer Res. 1981, 41, 1205–1208. [Google Scholar]
- Chang, B.K.; Libby, P.R.; Bergeron, R.J.; Porter, C.W. Modulation of polyamine biosynthesis and transport by oncogene transfection. Biochem. Biophys. Res. Commun. 1988, 157, 264–270. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Gerner, E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999, 5, 945–951. [Google Scholar] [PubMed]
- Seiler, N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 1. Selective enzyme inhibitors. Curr. Drug Targets 2003, 4, 537–564. [Google Scholar] [CrossRef] [PubMed]
- Sholler, G.L.S.; Gerner, E.W.; Bergendahl, G.; MacArthur, R.B.; VanderWerff, A.; Ashikaga, T.; Bond, J.P.; Ferguson, W.; Roberts, W.; Wada, R.K.; et al. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS ONE 2015, 10, e0127246. [Google Scholar]
- Sholler, G.L.S.; Ferguson, W.; Bergendahl, G.; Bond, J.P.; Neville, K.; Eslin, D.; Brown, V.; Roberts, W.; Wada, R.K.; Oesterheld, J.; et al. Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci. Rep. 2018, 8, 14445. [Google Scholar] [CrossRef]
- Muth, A.; Madan, M.; Archer, J.J.; Ocampo, N.; Rodriguez, L.; Phanstiel, O., IV. Polyamine transport inhibitors: Design, synthesis, and combination therapies with difluoromethylornithine. J. Med. Chem. 2014, 57, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Mariner, K.; Donnelly, J.; Phanstiel, O., 4th; Gilmour, S.K. Polyamine Blocking Therapy Decreases Survival of Tumor-Infiltrating Immunosuppressive Myeloid Cells and Enhances the Anti-Tumor Efficacy of PD-1 Blockade. Mol. Cancer Ther. 2020, 19, 2012–2022. [Google Scholar] [CrossRef]
- Ying, W.; Cheruku, P.S.; Bazer, F.W.; Safe, S.H.; Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp. 2013, 23, 50323. [Google Scholar]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. The Dawn of Myeloid-Derived Suppressor Cells: Identification of Arginase I as the Mechanism of Immune Suppression. Cancer Res. 2021, 81, 3953–3955. [Google Scholar] [CrossRef]
- Weber, R.; Riester, Z.; Huser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J. Immunol. 2005, 174, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.; Hernandez, C.P.; Raber, P.; Del Valle, L.; Wilk, A.M.; Majumdar, S.; Wyczechowska, D.; Reiss, K.; Rodriguez, P.C. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 2013, 27, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef]
- Parker, K.H.; Horn, L.A.; Ostrand-Rosenberg, S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J. Leukoc. Biol. 2016, 100, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lei, L.; Qin, W.; Wang, L.; Zhang, G.; Hu, J. GCN2 controls the cellular checkpoint: Potential target for regulating inflammation. Cell Death Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Ngabire, D.; Kim, G.D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexander, E.T.; Fahey, E.; Phanstiel, O., IV; Gilmour, S.K. Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice. Biomedicines 2023, 11, 2703. https://doi.org/10.3390/biomedicines11102703
Alexander ET, Fahey E, Phanstiel O IV, Gilmour SK. Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice. Biomedicines. 2023; 11(10):2703. https://doi.org/10.3390/biomedicines11102703
Chicago/Turabian StyleAlexander, Eric T., Erin Fahey, Otto Phanstiel, IV, and Susan K. Gilmour. 2023. "Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice" Biomedicines 11, no. 10: 2703. https://doi.org/10.3390/biomedicines11102703
APA StyleAlexander, E. T., Fahey, E., Phanstiel, O., IV, & Gilmour, S. K. (2023). Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice. Biomedicines, 11(10), 2703. https://doi.org/10.3390/biomedicines11102703