
Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Lines and Culture Conditions
2.2. Mouse Models
2.3. Reagents
2.4. Biodistribution Studies in Mice
2.5. LC–MS/MS Analyses of Tumors
2.6. Epichaperome Determination by Native PAGE
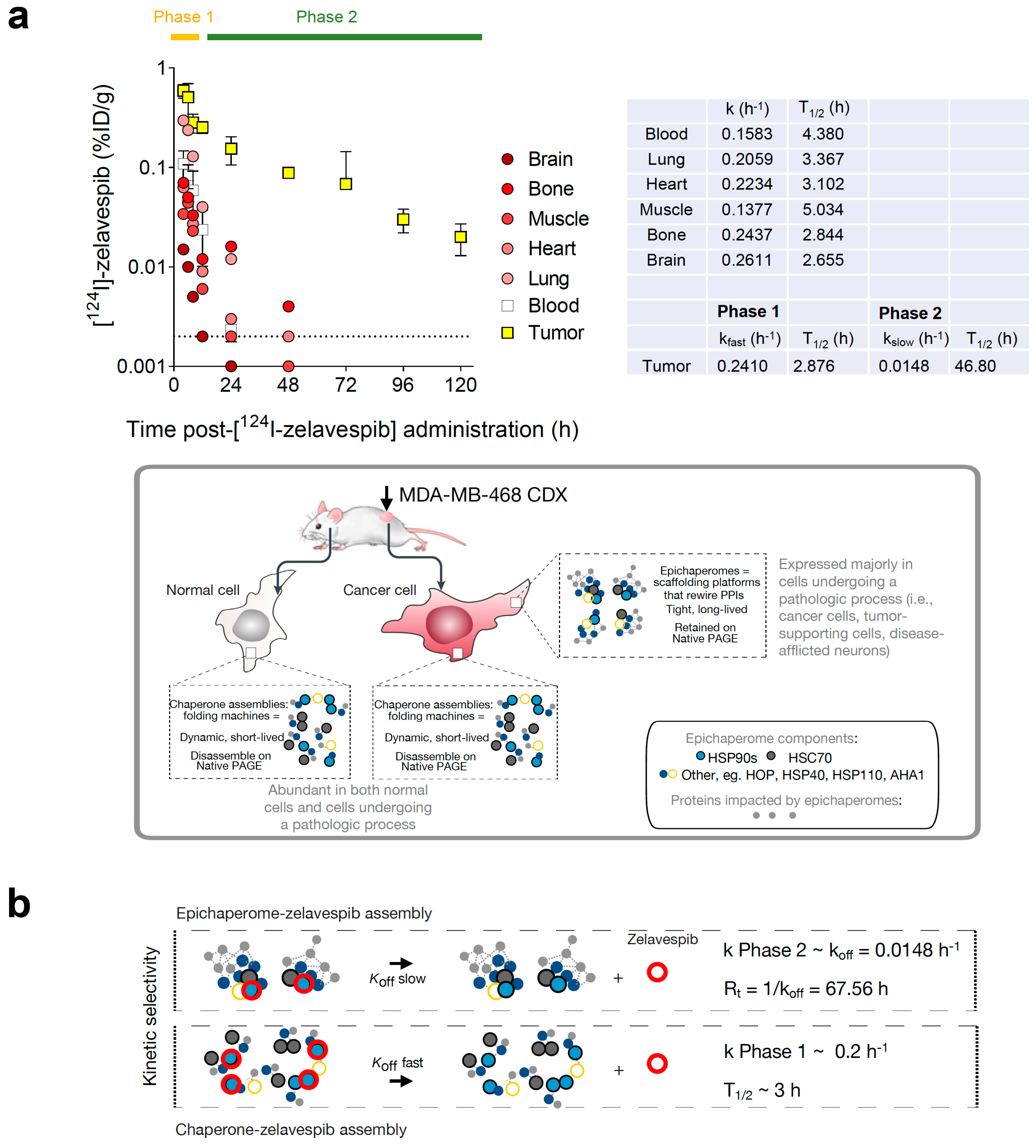
2.7. Use of Microdose [131I]-PU-AD Co-Injected with a Therapeutic Dose of Zelavespib or [124I]-PU-AD Injected Alone to Predict Tumor Molar Concentrations of Zelavespib
2.8. Statistics and Reproducibility
3. Results
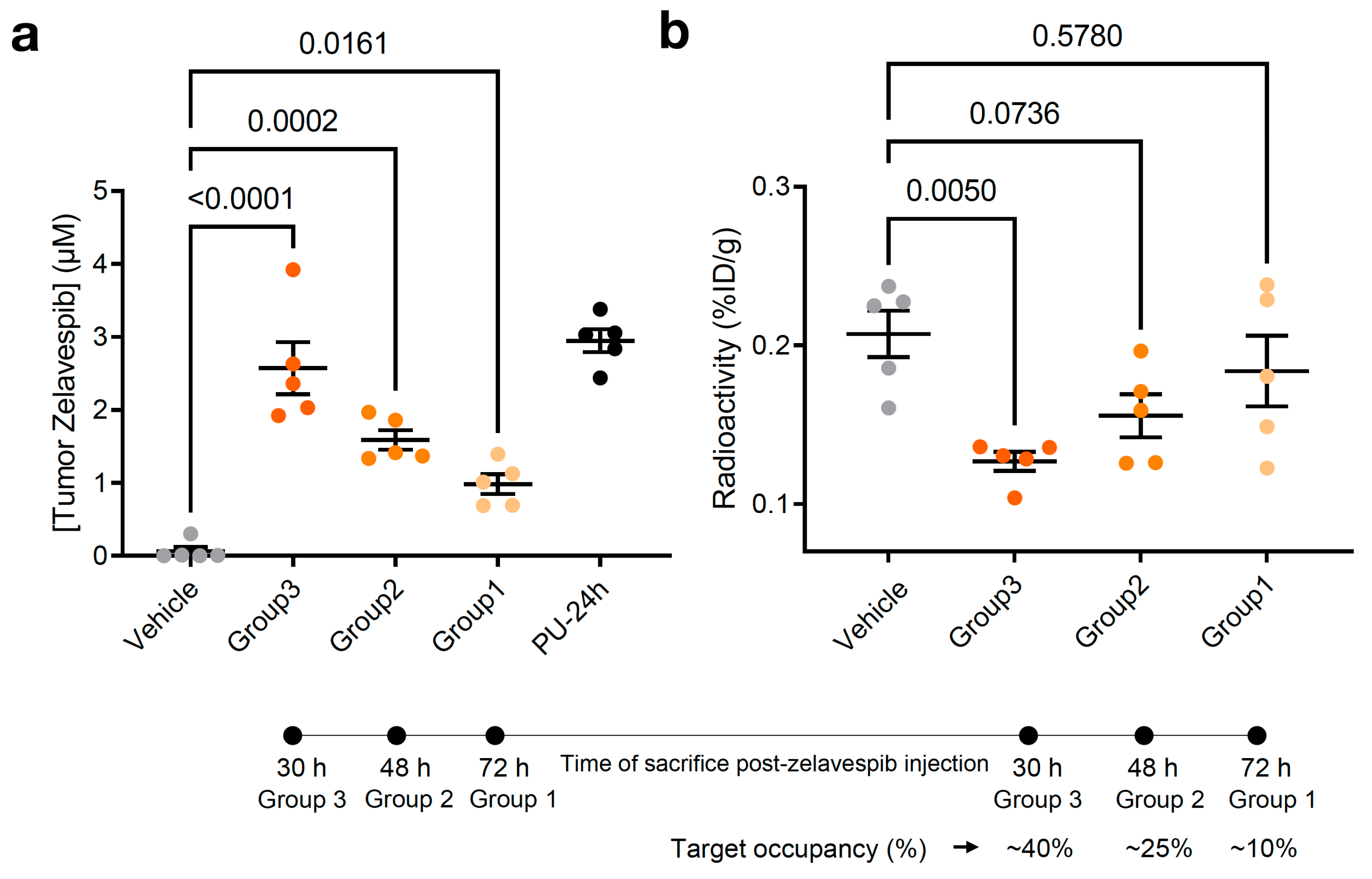
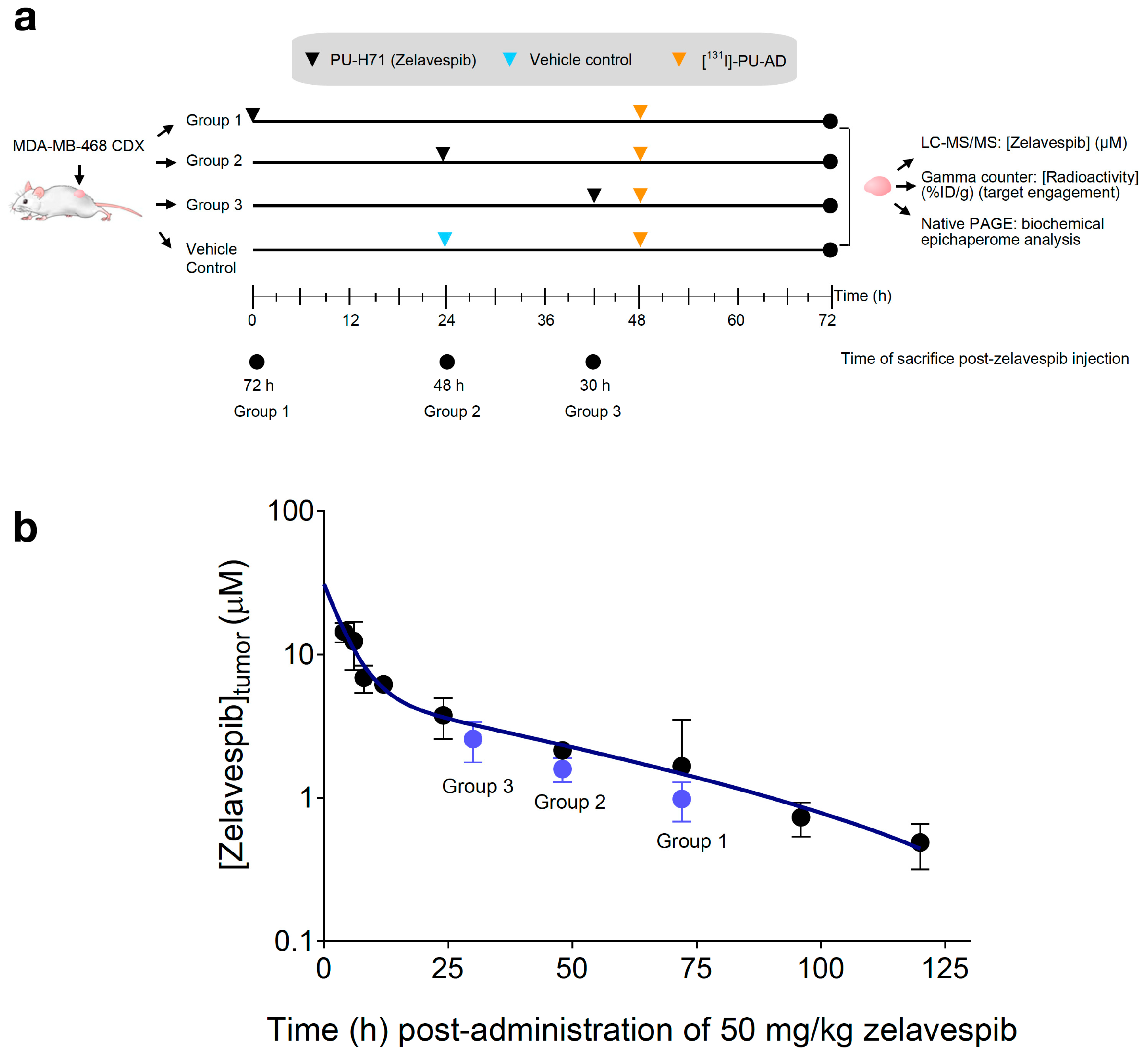
3.1. Experimental Design
3.2. Tumor Zelavespib Molar Concentration Measurements
3.3. Target Occupancy by Zelavespib
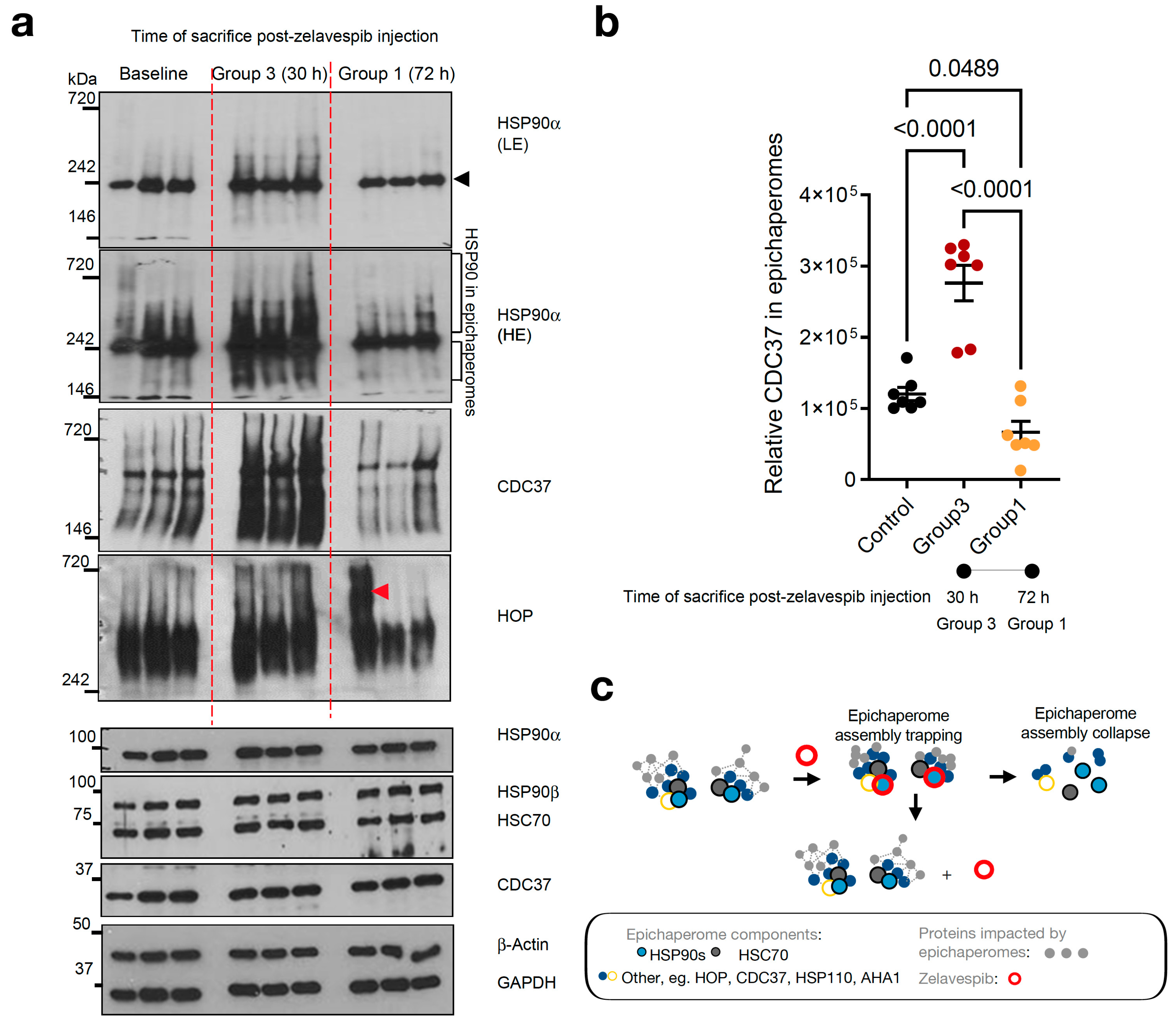
3.4. Modulation of Epichaperomes by Zelavespib
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Morgan, P.; Van Der Graaf, P.H.; Arrowsmith, J.; Feltner, D.E.; Drummond, K.S.; Wegner, C.D.; Street, S.D. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov. Today 2012, 17, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Markman, M. Drugs Can–t Kill a Tumor They Can–t Reach. Medscape, 6 February 2013. Available online: https://www.medscape.com/viewarticle/778630 (accessed on 7 July 2023).
- Emmerich, C.H.; Gamboa, L.M.; Hofmann, M.C.J.; Bonin-Andresen, M.; Arbach, O.; Schendel, P.; Gerlach, B.; Hempel, K.; Bespalov, A.; Dirnagl, U.; et al. Improving target assessment in biomedical research: The GOT-IT recommendations. Nat. Rev. Drug Discov. 2021, 20, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, J.; Hjorth, S. Turn On, Tune In, Turnover! Target Biology Impacts In Vivo Potency, Efficacy, and Clearance. Pharmacol. Rev. 2023, 75, 416–462. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, J.; Peletier, L.A.; Hjorth, S. In vivo potency revisited—Keep the target in sight. Pharmacol. Ther. 2018, 184, 177–188. [Google Scholar] [CrossRef]
- Guzzetti, S.; Morentin Gutierrez, P. An integrated modelling approach for targeted degradation: Insights on optimization, data requirements and PKPD predictions from semi- or fully-mechanistic models and exact steady state solutions. J. Pharmacokinet. Pharmacodyn. 2023, 50, 327–349. [Google Scholar] [CrossRef]
- Ganotra, G.K.; Wade, R.C. Prediction of Drug-Target Binding Kinetics by Comparative Binding Energy Analysis. ACS Med. Chem. Lett. 2018, 9, 1134–1139. [Google Scholar] [CrossRef]
- Knockenhauer, K.E.; Copeland, R.A. The importance of binding kinetics and drug-target residence time in pharmacology. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug-target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Dahl, G.; Akerud, T. Pharmacokinetics and the drug-target residence time concept. Drug Discov. Today 2013, 18, 697–707. [Google Scholar] [CrossRef]
- Georgi, V.; Andres, D.; Fernandez-Montalvan, A.E.; Stegmann, C.M.; Becker, A.; Mueller-Fahrnow, A. Binding kinetics in drug discovery—A current perspective. Front. Biosci. (Landmark Ed.) 2017, 22, 21–47. [Google Scholar] [CrossRef]
- Guo, D.; Hillger, J.M.; AP, I.J.; Heitman, L.H. Drug-target residence time—A case for G protein-coupled receptors. Med. Res. Rev. 2014, 34, 856–892. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. The role of binding kinetics in therapeutically useful drug action. Curr. Opin. Drug Discov. Devel. 2009, 12, 31–39. [Google Scholar] [PubMed]
- Vauquelin, G. Effects of target binding kinetics on in vivo drug efficacy: Koff, kon and rebinding. Br. J. Pharmacol. 2016, 173, 2319–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Monsma, F. Binding kinetics and mechanism of action: Toward the discovery and development of better and best in class drugs. Expert. Opin. Drug Discov. 2010, 5, 1023–1029. [Google Scholar] [CrossRef]
- Song, Y.; Jeong, H.; Kim, S.R.; Ryu, Y.; Baek, J.; Kwon, J.; Cho, H.; Kim, K.N.; Lee, J.J. Dissecting the impact of target-binding kinetics of protein binders on tumor localization. iScience 2021, 24, 102104. [Google Scholar] [CrossRef]
- Guo, D.; Heitman, L.H.; IJzerman, A.P. The Added Value of Assessing Ligand-Receptor Binding Kinetics in Drug Discovery. ACS Med. Chem. Lett. 2016, 7, 819–821. [Google Scholar] [CrossRef]
- de Witte, W.E.A.; Danhof, M.; van der Graaf, P.H.; de Lange, E.C.M. In vivo Target Residence Time and Kinetic Selectivity: The Association Rate Constant as Determinant. Trends Pharmacol. Sci. 2016, 37, 831–842. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Sharma, S.; Norton, L.; Chiosis, G. Targeting stressor-induced dysfunctions in protein-protein interaction networks via epichaperomes. Trends Pharmacol. Sci. 2023, 44, 20–33. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Joshi, S.; Sharma, S.; Guzman, G.; Wang, T.; Arancio, O.; Chiosis, G. The penalty of stress—Epichaperomes negatively reshaping the brain in neurodegenerative disorders. J. Neurochem. 2021, 159, 958–979. [Google Scholar] [CrossRef]
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2023, 1–19. [Google Scholar] [CrossRef]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsky, J.L.; Chiosis, G. Adapting to stress—Chaperome networks in cancer. Nat. Rev. Cancer 2018, 18, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Kishinevsky, S.; Wang, T.; Rodina, A.; Chung, S.Y.; Xu, C.; Philip, J.; Taldone, T.; Joshi, S.; Alpaugh, M.L.; Bolaender, A.; et al. HSP90-incorporating chaperome networks as biosensor for disease-related pathways in patient-specific midbrain dopamine neurons. Nat. Commun. 2018, 9, 4345. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.C.; Joshi, S.; Wang, T.; Bolaender, A.; Gandu, S.; Koren Iii, J.; Che, A.Y.; Taldone, T.; Yan, P.; Sun, W.; et al. The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun. 2020, 11, 319. [Google Scholar] [CrossRef]
- Yan, P.; Patel, H.J.; Sharma, S.; Corben, A.; Wang, T.; Panchal, P.; Yang, C.; Sun, W.; Araujo, T.L.; Rodina, A.; et al. Molecular Stressors Engender Protein Connectivity Dysfunction through Aberrant N-Glycosylation of a Chaperone. Cell Rep. 2020, 31, 107840. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Gomes, E.D.; Wang, T.; Corben, A.; Taldone, T.; Gandu, S.; Xu, C.; Sharma, S.; Buddaseth, S.; Yan, P.; et al. Pharmacologically controlling protein-protein interactions through epichaperomes for therapeutic vulnerability in cancer. Commun. Biol. 2021, 4, 1333. [Google Scholar] [CrossRef]
- Rickner, H.D.; Jiang, L.; Hong, R.; O’Neill, N.K.; Mojica, C.A.; Snyder, B.J.; Zhang, L.; Shaw, D.; Medalla, M.; Wolozin, B.; et al. Single cell transcriptomic profiling of a neuron-astrocyte assembloid tauopathy model. Nat. Commun. 2022, 13, 6275. [Google Scholar] [CrossRef]
- Rodina, A.; Xu, C.; Digwal, C.S.; Joshi, S.; Patel, Y.; Santhaseela, A.R.; Bay, S.; Merugu, S.; Alam, A.; Yan, P.; et al. Systems-level analyses of protein-protein interaction network dysfunctions via epichaperomics identify cancer-specific mechanisms of stress adaptation. Nat. Commun. 2023, 14, 3742. [Google Scholar] [CrossRef]
- Bolaender, A.; Zatorska, D.; He, H.; Joshi, S.; Sharma, S.; Digwal, C.S.; Patel, H.J.; Sun, W.; Imber, B.S.; Ochiana, S.O.; et al. Chemical tools for epichaperome-mediated interactome dysfunctions of the central nervous system. Nat. Commun. 2021, 12, 4669. [Google Scholar] [CrossRef]
- Castelli, M.; Yan, P.; Rodina, A.; Digwal, C.S.; Panchal, P.; Chiosis, G.; Moroni, E.; Colombo, G. How aberrant N-glycosylation can alter protein functionality and ligand binding: An atomistic view. Structure 2023, 31, 987–1004.e8. [Google Scholar] [CrossRef]
- Pillarsetty, N.; Jhaveri, K.; Taldone, T.; Caldas-Lopes, E.; Punzalan, B.; Joshi, S.; Bolaender, A.; Uddin, M.M.; Rodina, A.; Yan, P.; et al. Paradigms for Precision Medicine in Epichaperome Cancer Therapy. Cancer Cell 2019, 36, 559–573.e557. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Zhong, Y.; Sanborn, M.A.; Teoh, T.C.; Ruan, J.; Yusof, R.; Hang, J.; Henderson, M.J.; Fang, S. Small molecule grp94 inhibitors block dengue and Zika virus replication. Antivir. Res. 2019, 171, 104590. [Google Scholar] [CrossRef] [PubMed]
- Chaumonnot, K.; Masson, S.; Sikner, H.; Bouchard, A.; Baverel, V.; Bellaye, P.S.; Collin, B.; Garrido, C.; Kohli, E. The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death Dis. 2021, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, T.; Nakata, M.; Nagase, S.; Takahara, Y.; Honda-Ogawa, M.; Mori, Y.; Akamatsu, Y.; Yamaguchi, M.; Okamoto, S.; Kawabata, S. GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection. mBio 2021, 12, e0326920. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Dos Anjos, C.H.; Taldone, T.; Wang, R.; Comen, E.; Fornier, M.; Bromberg, J.F.; Ma, W.; Patil, S.; Rodina, A.; et al. Measuring Tumor Epichaperome Expression Using [(124)I] PU-H71 Positron Emission Tomography as a Biomarker of Response for PU-H71 Plus Nab-Paclitaxel in HER2-Negative Metastatic Breast Cancer. JCO Precis. Oncol. 2020, 4, 1414–1424. [Google Scholar] [CrossRef]
- Sugita, M.; Wilkes, D.C.; Bareja, R.; Eng, K.W.; Nataraj, S.; Jimenez-Flores, R.A.; Yan, L.; De Leon, J.P.; Croyle, J.A.; Kaner, J.; et al. Targeting the epichaperome as an effective precision medicine approach in a novel PML-SYK fusion acute myeloid leukemia. NPJ Precis. Oncol. 2021, 5, 44. [Google Scholar] [CrossRef]
- Silverman, M.H.; Duggan, S.; Bardelli, G.; Sadler, B.; Key, C.; Medlock, M.; Reynolds, L.; Wallner, B. Safety, Tolerability and Pharmacokinetics of Icapamespib, a Selective Epichaperome Inhibitor, in Healthy Adults. J. Prev. Alzheimers Dis. 2022, 9, 635–645. [Google Scholar] [CrossRef]
- Dunphy, M.P.S.; Pressl, C.; Pillarsetty, N.; Grkovski, M.; Modi, S.; Jhaveri, K.; Norton, L.; Beattie, B.J.; Zanzonico, P.B.; Zatorska, D.; et al. First-in-Human Trial of Epichaperome-Targeted PET in Patients with Cancer. Clin. Cancer Res. 2020, 26, 5178–5187. [Google Scholar] [CrossRef]
- Cailleau, R.; Olive, M.; Cruciger, Q.V. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. In Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef]
- He, H.; Zatorska, D.; Kim, J.; Aguirre, J.; Llauger, L.; She, Y.; Wu, N.; Immormino, R.M.; Gewirth, D.T.; Chiosis, G. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J. Med. Chem. 2006, 49, 381–390. [Google Scholar] [CrossRef]
- Taldone, T.; Zatorska, D.; Kang, Y.; Chiosis, G. A facile and efficient synthesis of d6-labeled PU-H71, a purine-scaffold Hsp90 inhibitor. J. Label. Compd. Radiopharm. 2010, 53, 47–49. [Google Scholar] [CrossRef]
- Sharma, S.; Kalidindi, T.; Joshi, S.; Digwal, C.S.; Panchal, P.; Burnazi, E.; Lee, S.G.; Pillarsetty, N.; Chiosis, G. Synthesis of (124)I-labeled epichaperome probes and assessment in visualizing pathologic protein-protein interaction networks in tumor bearing mice. STAR Protoc. 2022, 3, 101318. [Google Scholar] [CrossRef] [PubMed]
- Speranza, G.; Anderson, L.; Chen, A.P.; Do, K.; Eugeni, M.; Weil, M.; Rubinstein, L.; Majerova, E.; Collins, J.; Horneffer, Y.; et al. First-in-human study of the epichaperome inhibitor PU-H71: Clinical results and metabolic profile. Investig. New Drugs 2018, 36, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Vauquelin, G.; Huber, W.; Swinney, D.C. Experimental Methods to Determine Binding Kinetics. In Thermodynamics and Kinetics of Drug Binding; Wiley-VCH Verlag GmbH & Co. KGaA: Wernheim, Germany, 2015; pp. 169–189. [Google Scholar]
- Robers, M.B.; Vasta, J.D.; Corona, C.R.; Ohana, R.F.; Hurst, R.; Jhala, M.A.; Comess, K.M.; Wood, K.V. Quantitative, Real-Time Measurements of Intracellular Target Engagement Using Energy Transfer. Methods Mol. Biol. 2019, 1888, 45–71. [Google Scholar] [CrossRef]
- Mettler, F.A.; Guiberteau, M.J. Essentials of Nuclear Medicine Imaging E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Ehrhardt, J.D., Jr.; Gulec, S. A Review of the History of Radioactive Iodine Theranostics: The Origin of Nuclear Ontology. Mol. Imaging Radionucl. Ther. 2020, 29, 88–97. [Google Scholar] [CrossRef]
- Woodson, S.A.; Koculi, E. Analysis of RNA folding by native polyacrylamide gel electrophoresis. Methods Enzymol. 2009, 469, 189–208. [Google Scholar] [CrossRef]
- Woodford, M.R.; Bourboulia, D.; Mollapour, M. Epichaperomics reveals dysfunctional chaperone protein networks. Nat. Commun. 2023, 14, 5084. [Google Scholar] [CrossRef]
- Gebre, S.T.; Cameron, S.A.; Li, L.; Babu, Y.S.; Schramm, V.L. Intracellular rebinding of transition-state analogues provides extended in vivo inhibition lifetimes on human purine nucleoside phosphorylase. J. Biol. Chem. 2017, 292, 15907–15915. [Google Scholar] [CrossRef]
- Lee, K.S.S.; Yang, J.; Niu, J.; Ng, C.J.; Wagner, K.M.; Dong, H.; Kodani, S.D.; Wan, D.; Morisseau, C.; Hammock, B.D. Drug-Target Residence Time Affects in Vivo Target Occupancy through Multiple Pathways. ACS Cent. Sci. 2019, 5, 1614–1624. [Google Scholar] [CrossRef]
- Vauquelin, G.; Charlton, S.J. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 2010, 161, 488–508. [Google Scholar] [CrossRef]
- Bosma, R.; Witt, G.; Vaas, L.A.I.; Josimovic, I.; Gribbon, P.; Vischer, H.F.; Gul, S.; Leurs, R. The Target Residence Time of Antihistamines Determines Their Antagonism of the G Protein-Coupled Histamine H1 Receptor. Front. Pharmacol. 2017, 8, 667. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Bradley, M.E.; Riddy, D.M.; Willard, E.; Reilly, J.; Miah, A.; Bauer, C.; Watson, S.J.; Sandham, D.A.; Dubois, G.; et al. Fevipiprant (QAW039), a Slowly Dissociating CRTh2 Antagonist with the Potential for Improved Clinical Efficacy. Mol. Pharmacol. 2016, 89, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Sachs, G. Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep. 2008, 10, 528–534. [Google Scholar] [CrossRef]
- Calcaterra, N.E.; Barrow, J.C. Classics in chemical neuroscience: Diazepam (valium). ACS Chem. Neurosci. 2014, 5, 253–260. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; IJzerman, A.P.; Guo, D. The translational perspective of ligand-receptor binding kinetics in drug discovery. Br. J. Pharmacol. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Tonge, P.J. Drug-Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9, 29–39. [Google Scholar] [CrossRef]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898. [Google Scholar] [CrossRef]
- Georgi, V.; Schiele, F.; Berger, B.T.; Steffen, A.; Marin Zapata, P.A.; Briem, H.; Menz, S.; Preusse, C.; Vasta, J.D.; Robers, M.B.; et al. Binding Kinetics Survey of the Drugged Kinome. J. Am. Chem. Soc. 2018, 140, 15774–15782. [Google Scholar] [CrossRef]
- Hoare, S.R.J.; Fleck, B.A.; Williams, J.P.; Grigoriadis, D.E. The importance of target binding kinetics for measuring target binding affinity in drug discovery: A case study from a CRF(1) receptor antagonist program. Drug Discov. Today 2020, 25, 7–14. [Google Scholar] [CrossRef]
- Hothersall, J.D.; Brown, A.J.; Dale, I.; Rawlins, P. Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discov. Today 2016, 21, 90–96. [Google Scholar] [CrossRef]
- Lu, H.; Tonge, P.J. Drug-target residence time: Critical information for lead optimization. Curr. Opin. Chem. Biol. 2010, 14, 467–474. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Joshi, S.; Kalidindi, T.; Digwal, C.S.; Panchal, P.; Lee, S.-G.; Zanzonico, P.; Pillarsetty, N.; Chiosis, G. Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action. Biomedicines 2023, 11, 2599. https://doi.org/10.3390/biomedicines11102599
Sharma S, Joshi S, Kalidindi T, Digwal CS, Panchal P, Lee S-G, Zanzonico P, Pillarsetty N, Chiosis G. Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action. Biomedicines. 2023; 11(10):2599. https://doi.org/10.3390/biomedicines11102599
Chicago/Turabian StyleSharma, Sahil, Suhasini Joshi, Teja Kalidindi, Chander S. Digwal, Palak Panchal, Sang-Gyu Lee, Pat Zanzonico, Nagavarakishore Pillarsetty, and Gabriela Chiosis. 2023. "Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action" Biomedicines 11, no. 10: 2599. https://doi.org/10.3390/biomedicines11102599
APA StyleSharma, S., Joshi, S., Kalidindi, T., Digwal, C. S., Panchal, P., Lee, S.-G., Zanzonico, P., Pillarsetty, N., & Chiosis, G. (2023). Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action. Biomedicines, 11(10), 2599. https://doi.org/10.3390/biomedicines11102599