
Integrin β1/Cell Surface GRP78 Complex Regulates TGFβ1 and Its Profibrotic Effects in Response to High Glucose

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Reagents and Protein Extraction
2.2. Transfection and Luciferase
2.3. RNA Extraction and qPCR
2.4. Biotinylation
2.5. TGFβ1 ELISA
2.6. TGFβ1 Bioassay with Mink Lung Epithelial Cells (MLECs)
2.7. Surface Protein Co-Immunoprecipitation from Live Cells
2.8. Statistical Analysis
3. Results
3.1. csGRP78 Mediates TGFβ1 Transcription in Response to HG
3.2. HG-Induced TGFβ1 Protein Synthesis and Secretion Are Mediated by csGRP78
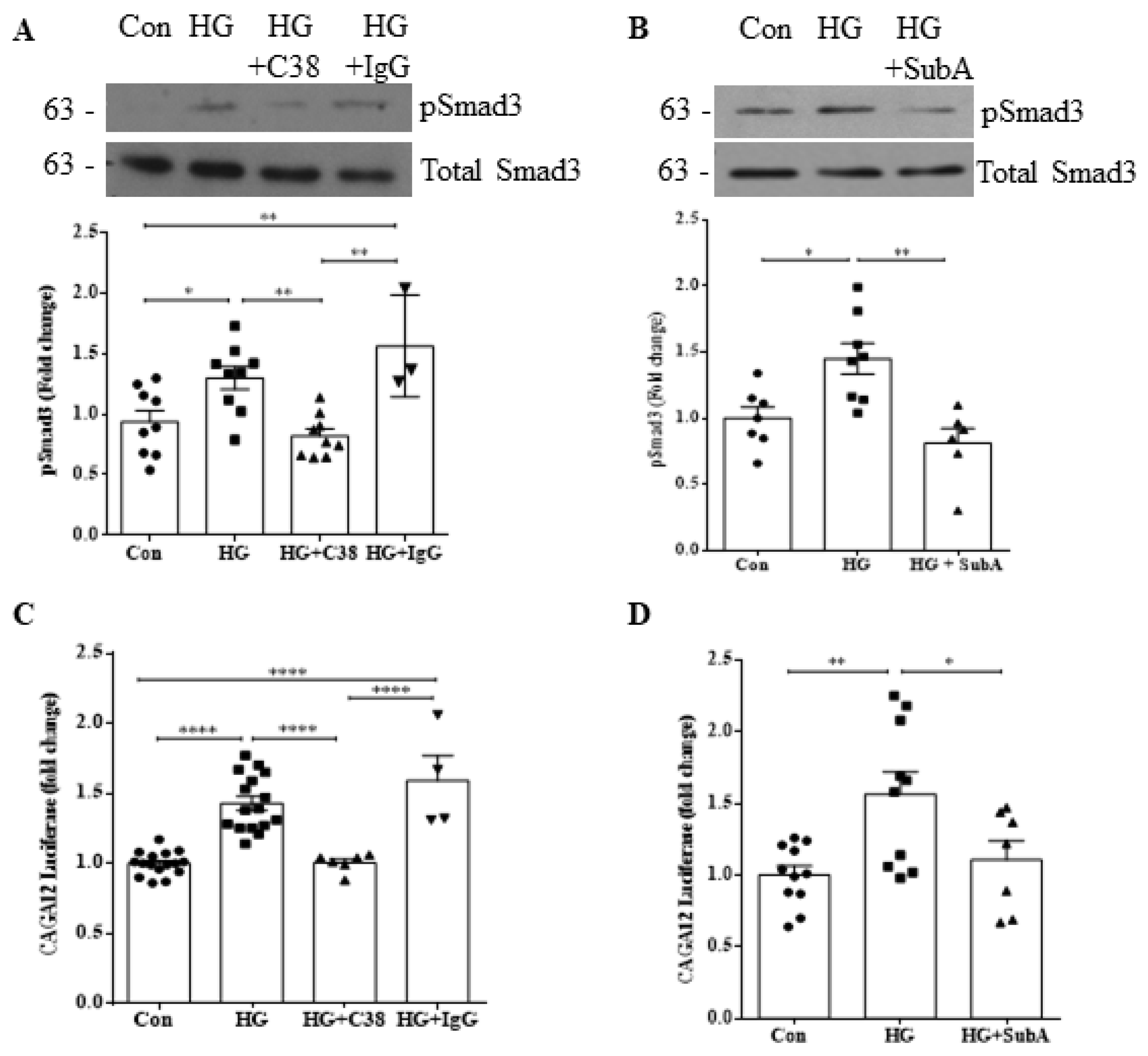
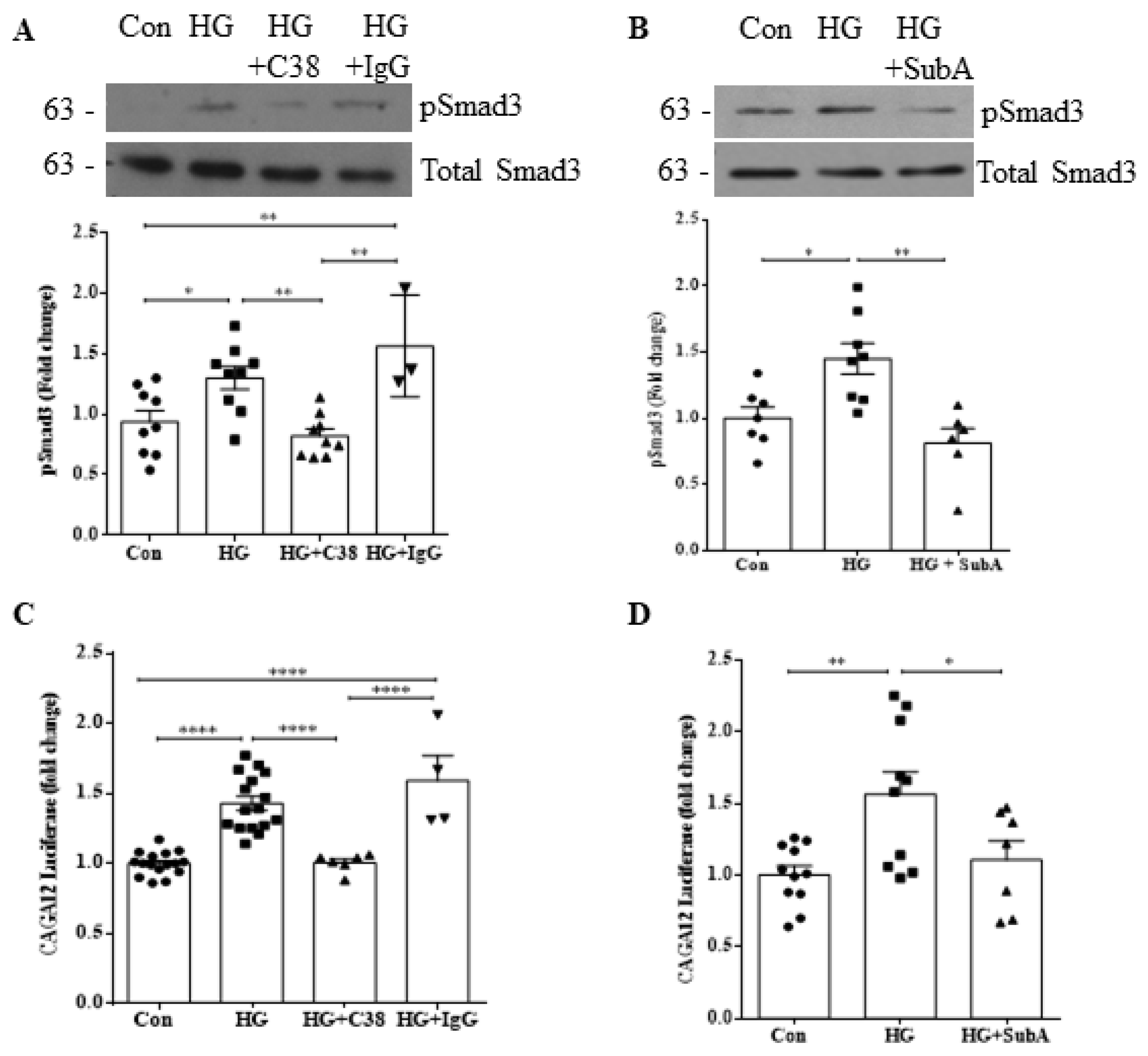
3.3. csGRP78 Facilitates HG-Induced Smad3 Activation
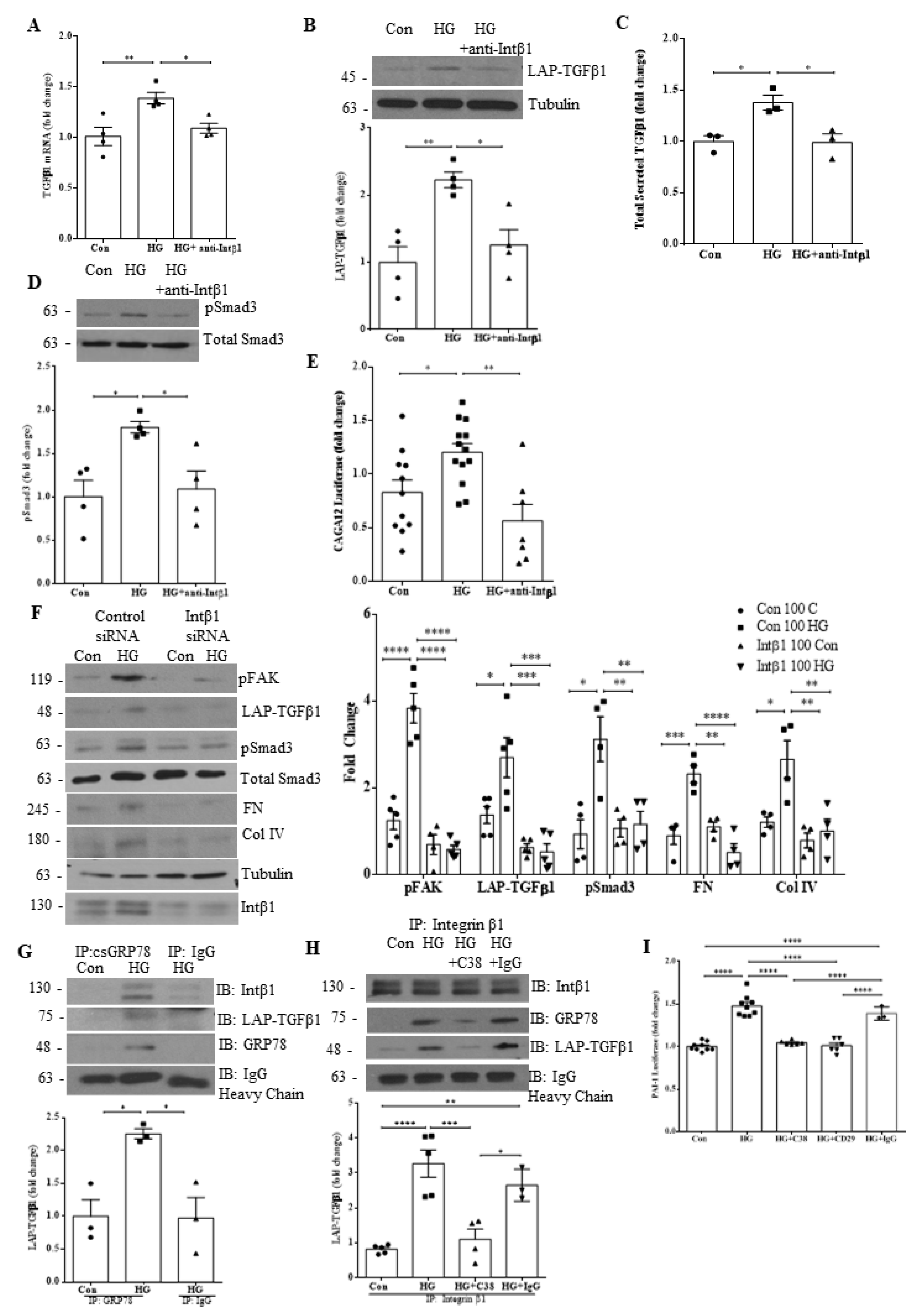
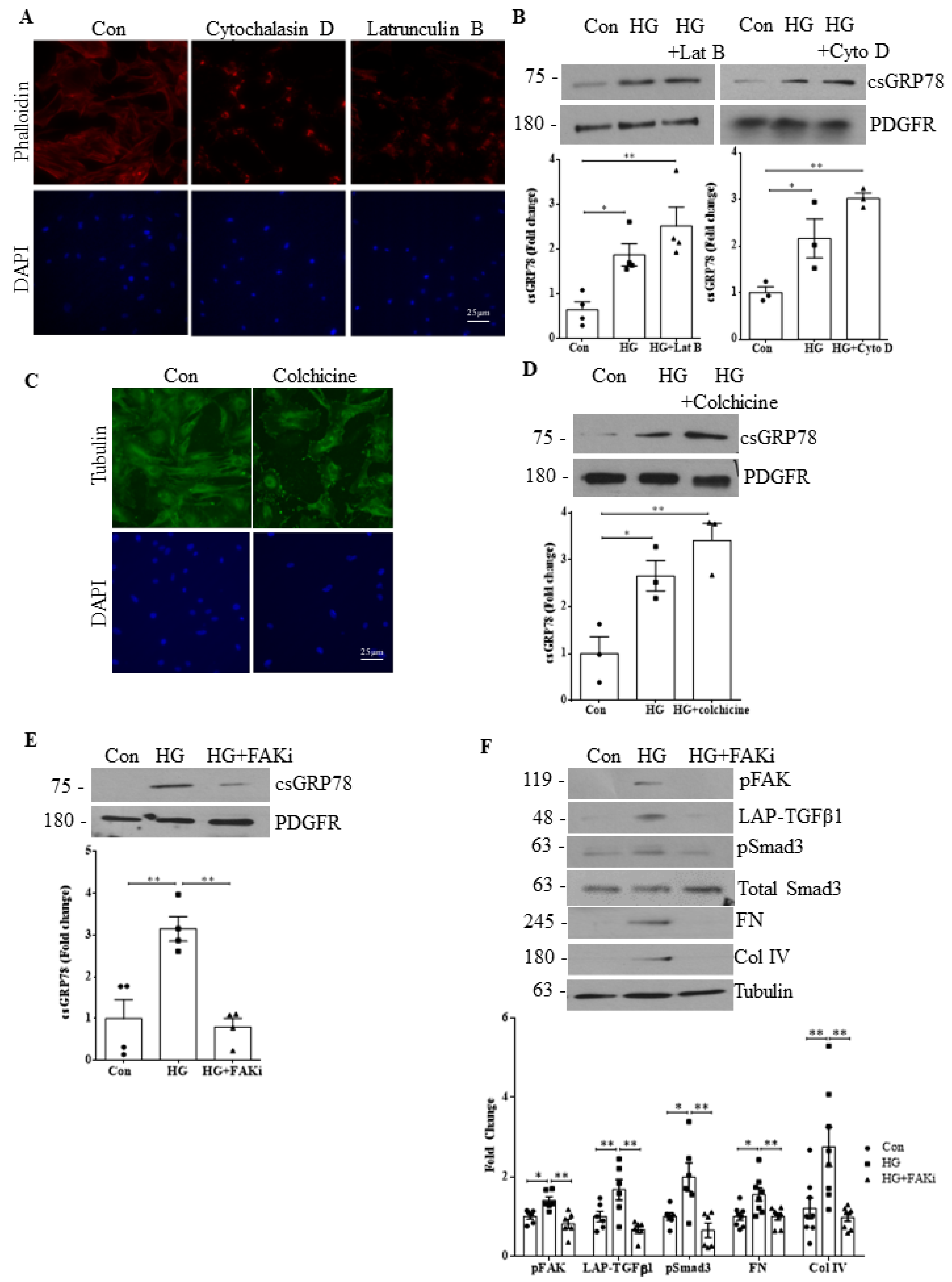
3.4. Integrin β1 Interaction with csGRP78 Is Required for TGFβ1 Upregulation and Signaling in HG
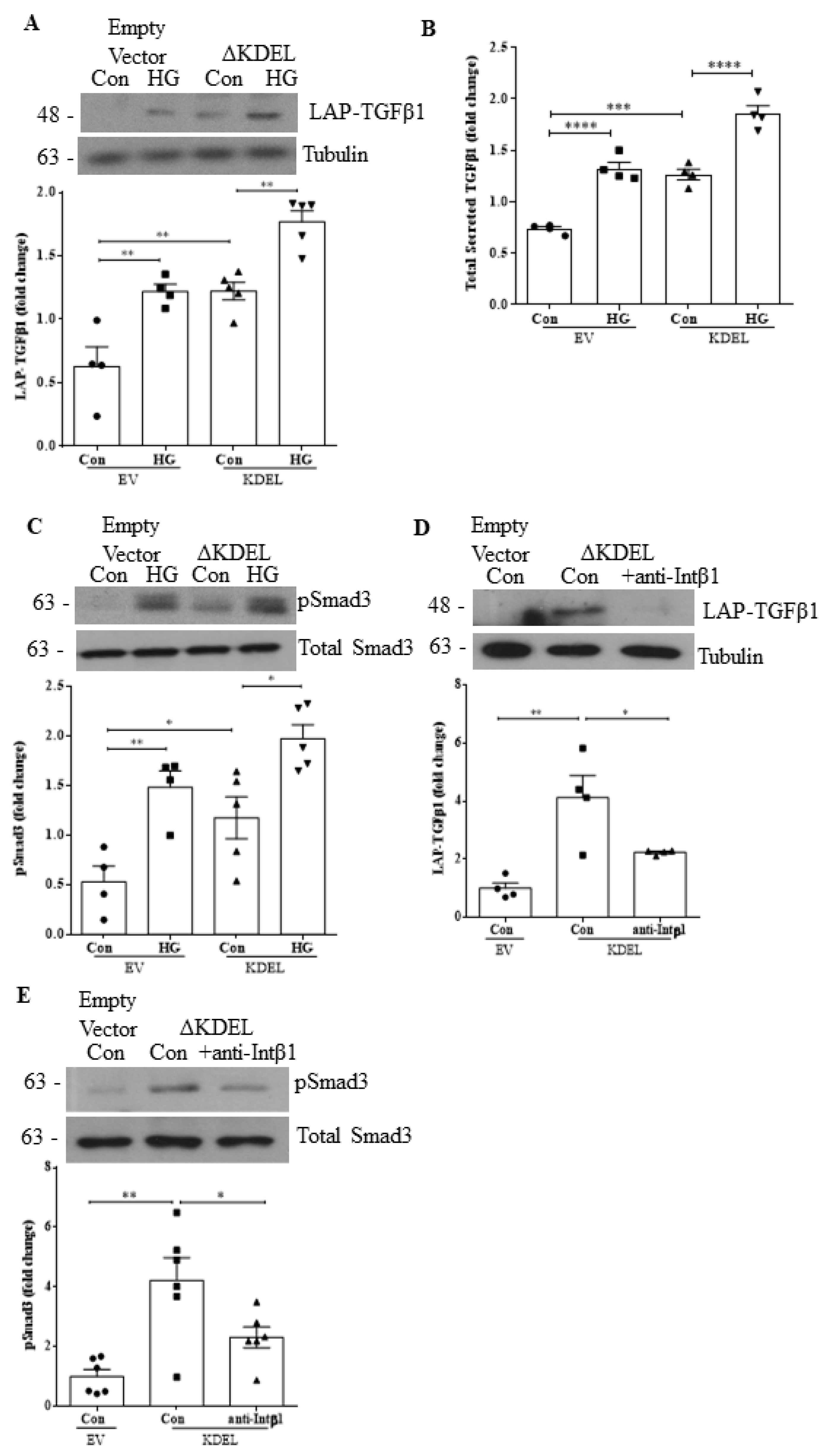
3.5. Overexpression of csGRP78 Augments TGFβ1 Synthesis, Secretion and Downstream Profibrotic Signaling
3.6. Integrin β1 Contributes to GRP78 Cell Surface Translocation
3.7. HG-Induced csGRP78 Translocation Is Independent of Cytoskeleton Organization
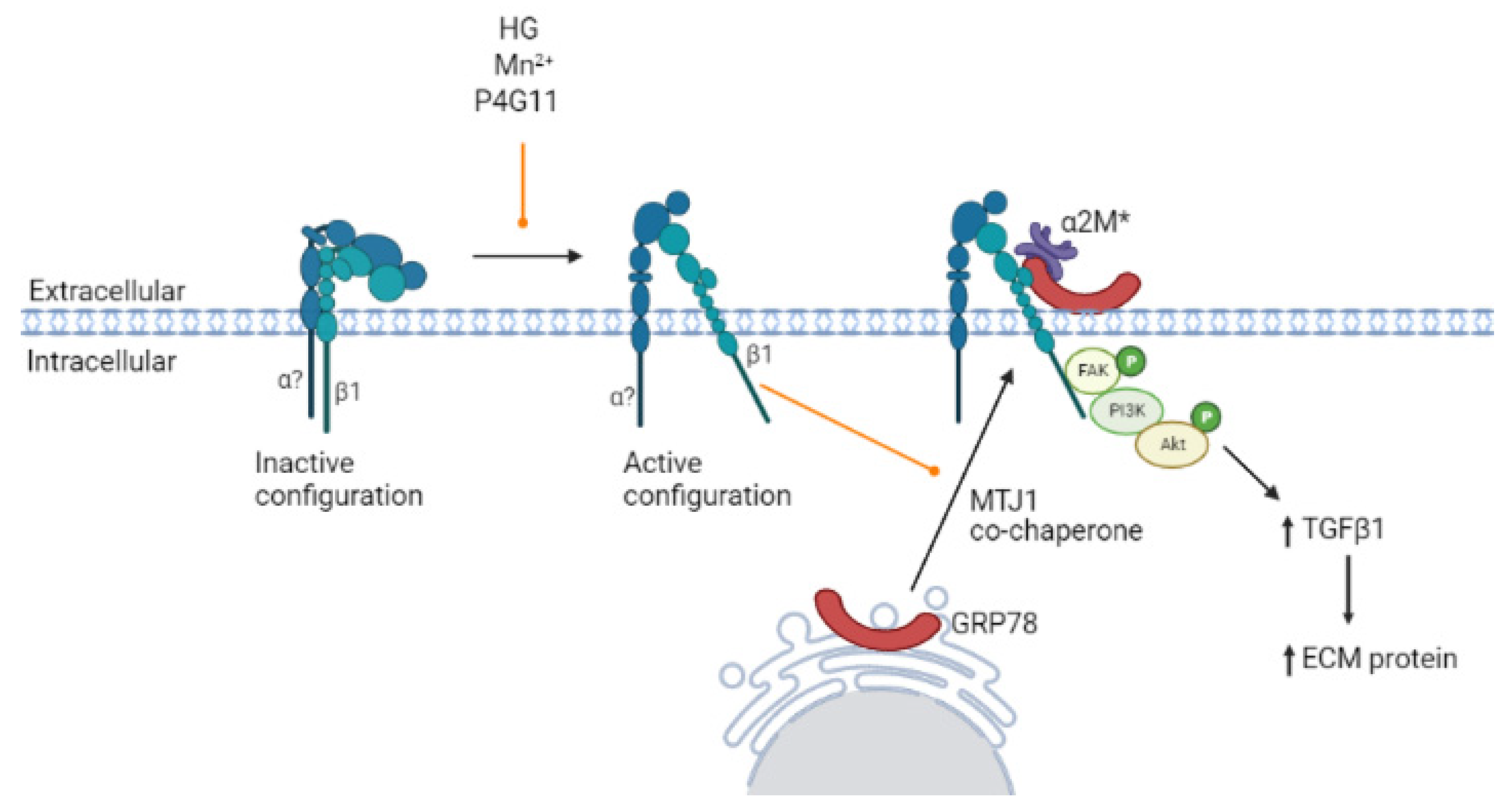
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Spurney, R.F. Twenty years after ACEIs and ARBs: Emerging treatment strategies for diabetic nephropathy. Am. J. Physiol. Physiol. 2015, 309, F807–F820. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C.; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 Inhibition for CKD and Cardiovascular Disease in Type 2 Diabetes: Report of a Scientific Workshop Sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 2021, 77, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Sagoo, M.K.; Gnudi, L. Diabetic Nephropathy: An Overview. Diabet. Nephrop. 2019, 2067, 3–7. [Google Scholar] [CrossRef]
- Warren, A.; Knudsen, S.T.; Cooper, M.E. Diabetic nephropathy: An insight into molecular mechanisms and emerging therapies. Expert Opin. Ther. Targets 2019, 23, 579–591. [Google Scholar] [CrossRef]
- Van Krieken, R.; Mehta, N.; Wang, T.; Zheng, M.; Li, R.; Gao, B.; Ayaub, E.; Ask, K.; Paton, J.C.; Paton, A.W.; et al. Cell surface expression of 78-kDa glucose-regulated protein (GRP78) mediates diabetic nephropathy. J. Biol. Chem. 2019, 294, 7755–7768. [Google Scholar] [CrossRef]
- Trink, J.; Li, R.; Palarasah, Y.; Troyanov, S.; Andersen, T.E.; Sidelmann, J.J.; Inman, M.D.; Pizzo, S.V.; Gao, B.; Krepinsky, J.C. Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease. Biomedicines 2021, 9, 1112. [Google Scholar] [CrossRef]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2014, 230, 1413–1420. [Google Scholar] [CrossRef]
- Gonzalez–Gronow, M.; Selim, M.A.; Papalas, J.; Pizzo, S.V. GRP78: A Multifunctional Receptor on the Cell Surface. Antioxid. Redox Signal. 2009, 11, 2299–2306. [Google Scholar] [CrossRef]
- Oida, T.; Weiner, H.L. Overexpression of TGF-β1Gene Induces Cell Surface Localized Glucose-Regulated Protein 78-Associated Latency-Associated Peptide/TGF-β. J. Immunol. 2010, 185, 3529–3535. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-L.; Zhang, Y.; Tseng, C.-C.; Stanciauskas, R.; Pinaud, F.; Lee, A.S. Characterization and Mechanism of Stress-induced Translocation of 78-Kilodalton Glucose-regulated Protein (GRP78) to the Cell Surface. J. Biol. Chem. 2015, 290, 8049–8064. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Kelly, D.J.; Gilbert, R.E.; Gao, B.; Krepinsky, J.C. PKC-β1 Mediates Glucose-Induced Akt Activation and TGF-β1 Upregulation in Mesangial Cells. J. Am. Soc. Nephrol. 2009, 20, 554–566. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.-J.; Johnson, R.; Lan, H.Y. Role of TGF-β signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003, 63, 2010–2019. [Google Scholar] [CrossRef]
- Ray, R.; de Ridder, G.G.; Eu, J.P.; Paton, A.W.; Paton, J.C.; Pizzo, S.V. The Escherichia coli Subtilase Cytotoxin A Subunit Specifically Cleaves Cell-surface GRP78 Protein and Abolishes COOH-terminal-dependent Signaling. J. Biol. Chem. 2012, 287, 32755–32769. [Google Scholar] [CrossRef]
- De Ridder, G.G.; Ray, R.; Pizzo, S.V. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012, 22, 225–235. [Google Scholar] [CrossRef]
- Krepinsky, J.C.; Ingram, A.J.; Tang, D.; Wu, D.; Liu, L.; Scholey, J.W. Nitric Oxide Inhibits Stretch-Induced MAPK Activation in Mesangial Cells Through RhoA Inactivation. J. Am. Soc. Nephrol. 2003, 14, 2790–2800. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Pizzo, S.V. The Role of MTJ-1 in Cell Surface Translocation of GRP78, a Receptor for α2-Macroglobulin-Dependent Signaling. J. Immunol. 2005, 174, 2092–2097. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Kamaraju, A.K.; Wakefield, L.M.; Roberts, A.B. Lentiviral reporter constructs for fluorescence tracking of the temporospatial pattern of Smad3 signaling. BioTechniques 2007, 43, 289–294. [Google Scholar] [CrossRef]
- Lin, G.L.; Cohen, D.M.; Desai, R.A.; Breckenridge, M.T.; Gao, L.; Humphries, M.J.; Chen, C.S. Activation of beta 1 but not beta 3 integrin increases cell traction forces. FEBS Lett. 2013, 587, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Kwok, J.; Patani, R.; Ffrench-Constant, C.; Chandran, S.; Fawcett, J. Integrin Activation Promotes Axon Growth on Inhibitory Chondroitin Sulfate Proteoglycans by Enhancing Integrin Signaling. J. Neurosci. 2011, 31, 6289–6295. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Shattil, S.J.; Ginsberg, M.H. Integrins and Actin Filaments: Reciprocal Regulation of Cell Adhesion and Signaling. J. Biol. Chem. 2000, 275, 22607–22610. [Google Scholar] [CrossRef] [PubMed]
- Delon, I.; Brown, N.H. Integrins and the actin cytoskeleton. Curr. Opin. Cell Biol. 2007, 19, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Pozzi, A.; Zent, R. Integrins in Kidney Disease. J. Am. Soc. Nephrol. 2013, 24, 1034–1039. [Google Scholar] [CrossRef]
- Miller, C.G.; Pozzi, A.; Zent, R.; Schwarzbauer, J.E. Effects of high glucose on integrin activity and fibronectin matrix assembly by mesangial cells. Mol. Biol. Cell 2014, 25, 2342–2350. [Google Scholar] [CrossRef]
- Chang, Y.; Lau, W.L.; Jo, H.; Tsujino, K.; Gewin, L.; Reed, N.I.; Atakilit, A.; Nunes, A.C.F.; DeGrado, W.F.; Sheppard, D. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in Vivo. J. Am. Soc. Nephrol. 2017, 28, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Klementowicz, J.E.; Travis, M.A. TGFβ: A sleeping giant awoken by integrins. Trends Biochem. Sci. 2011, 36, 47–54. [Google Scholar] [CrossRef]
- Yevdokimova, N.; Wahab, N.A.; Mason, R.M. Thrombospondin-1 Is the Key Activator of TGF-β1 in Human Mesangial Cells Exposed to High Glucose. J. Am. Soc. Nephrol. 2001, 12, 703–712. [Google Scholar] [CrossRef]
- Daniel, C.; Schaub, K.; Amann, K.; Lawler, J.; Hugo, C. Thrombospondin-1 Is an Endogenous Activator of TGF-β in Experimental Diabetic Nephropathy In Vivo. Diabetes 2007, 56, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E. Thrombospondin 1 and Its Diverse Roles as a Regulator of Extracellular Matrix in Fibrotic Disease. J. Histochem. Cytochem. 2019, 67, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.R.; Owens, T.W.; Naylor, M.J. Structural and mechanical functions of integrins. Biophys. Rev. 2013, 6, 203–213. [Google Scholar] [CrossRef]
- Borza, C.M.; Su, Y.; Chen, X.; Yu, L.; Mont, S.; Chetyrkin, S.; Voziyan, P.; Hudson, B.G.; Billings, P.C.; Jo, H.; et al. Inhibition of Integrin α2β1 Ameliorates Glomerular Injury. J. Am. Soc. Nephrol. 2012, 23, 1027–1038. [Google Scholar] [CrossRef]
- Weston, B.S.; Wahab, N.A.; Mason, R.M. CTGF Mediates TGF-β–Induced Fibronectin Matrix Deposition by Upregulating Active α5β1 Integrin in Human Mesangial Cells. J. Am. Soc. Nephrol. 2003, 14, 601–610. [Google Scholar] [CrossRef]
- Kitsiou, P.V.; Tzinia, A.K.; Stetler-Stevenson, W.G.; Michael, A.F.; Fan, W.W.; Zhou, B.; Tsilibary, E.C. Glucose-induced changes in integrins and matrix-related functions in cultured human glomerular epi-thelial cells. Am. J. Physiol.-Ren. Physiol. 2003, 284, F671–F679. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zeng, Z.; Wu, T.; Yang, Z.; Liu, B.; Lan, T. Emodin attenuates high glucose-induced TGF-β1 and fibronectin expression in mesangial cells through inhibition of NF-κB pathway. Exp. Cell Res. 2013, 319, 3182–3189. [Google Scholar] [CrossRef] [PubMed]
- Maile, L.A.; Busby, W.H.; Gollahon, K.A.; Flowers, W.; Garbacik, N.; Garbacik, S.; Stewart, K.; Nichols, T.; Bellinger, D.; Patel, A.; et al. Blocking Ligand Occupancy of the αVβ3 Integrin Inhibits the Development of Nephropathy in Diabetic Pigs. Endocrinology 2014, 155, 4665–4675. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Gao, B.; Chen, G.; Chen, X.; Janssen, M.; Uttarwar, L.; Ingram, A.J.; Krepinsky, J.C. Colchicine attenuates renal injury in a model of hypertensive chronic kidney disease. Am. J. Physiol. Physiol. 2013, 305, F1466–F1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trink, J.; Li, R.; Squire, E.; O’Neil, K.; Zheng, P.; Gao, B.; Krepinsky, J.C. Integrin β1/Cell Surface GRP78 Complex Regulates TGFβ1 and Its Profibrotic Effects in Response to High Glucose. Biomedicines 2022, 10, 2247. https://doi.org/10.3390/biomedicines10092247
Trink J, Li R, Squire E, O’Neil K, Zheng P, Gao B, Krepinsky JC. Integrin β1/Cell Surface GRP78 Complex Regulates TGFβ1 and Its Profibrotic Effects in Response to High Glucose. Biomedicines. 2022; 10(9):2247. https://doi.org/10.3390/biomedicines10092247
Chicago/Turabian StyleTrink, Jackie, Renzhong Li, Evan Squire, Kian O’Neil, Phoebe Zheng, Bo Gao, and Joan C. Krepinsky. 2022. "Integrin β1/Cell Surface GRP78 Complex Regulates TGFβ1 and Its Profibrotic Effects in Response to High Glucose" Biomedicines 10, no. 9: 2247. https://doi.org/10.3390/biomedicines10092247
APA StyleTrink, J., Li, R., Squire, E., O’Neil, K., Zheng, P., Gao, B., & Krepinsky, J. C. (2022). Integrin β1/Cell Surface GRP78 Complex Regulates TGFβ1 and Its Profibrotic Effects in Response to High Glucose. Biomedicines, 10(9), 2247. https://doi.org/10.3390/biomedicines10092247