
Bioinformatics: From NGS Data to Biological Complexity in Variant Detection and Oncological Clinical Practice

 , and
, and 
Abstract
1. Introduction
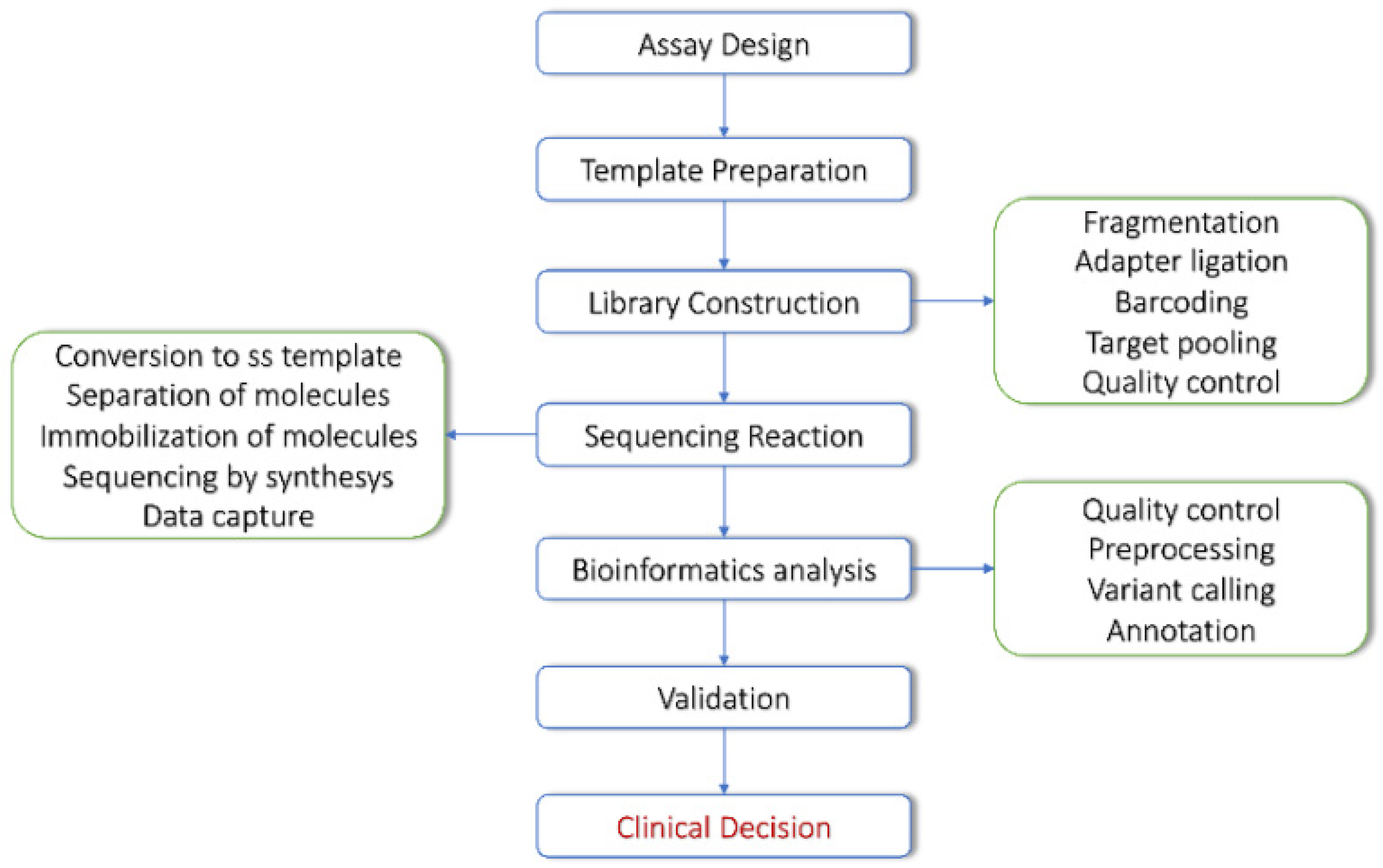
2. NGS Technologies and Bioinformatics Tools for DNA Sequencing Data
NGS Approaches in Cancer Patients’ Management
3. From NGS Data to Variant Discovery
3.1. Variant Types Relevant for Precision Oncology
3.2. Variant Discovery Workflow
3.3. Sequencing Quality Control
3.4. Pre Processing
3.5. Variant Calling
3.6. Variant Discovery Pipelines: Tools and Algorithms
3.7. Annotation
4. Machine Learning Applied to Next-Generation Sequencing and Variant Discovery
4.1. Sequencing Technology Issues and Machine Learning
4.2. ML and Deep Neural Networks for Variant Discovery
4.3. Machine Learning Development Frameworks
5. Network-Based Approaches Applied to Cancer Research: Graph Theory and Causality for Analyzing the Biological Complexity
5.1. Graph Theory
5.2. Causality
6. Homologous Recombination Deficiency: A New Bioinformatics Challenge
6.1. Testing Strategies for HRD Detection Based on Causes and Effects in the Genome
6.2. Computational Tools for HRD Assessment
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hussen, B.M.; Abdullah, S.T.; Salihi, A.; Sabir, D.K.; Sidiq, K.R.; Rasul, M.F.; Hidayat, H.J.; Ghafouri-Fard, S.; Taheri, M.; Jamali, E. The emerging roles of NGS in clinical oncology and personalized medicine. Pathol. Res. Pract. 2022, 230, 153760. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Steuten, L.; Aftimos, P.; André, F.; Davies, M.; Garralda, E.; Geissler, J.; Husereau, D.; Martinez-Lopez, I.; Normanno, N.; et al. Delivering precision oncology to patients with cancer. Nat. Med. 2022, 28, 658–665. [Google Scholar] [CrossRef]
- Normanno, N.; Apostolidis, K.; de Lorenzo, F.; Beer, P.A.; Henderson, R.; Sullivan, R.; Biankin, A.V.; Horgan, D.; Lawler, M. Cancer Biomarkers in the era of precision oncology: Addressing the needs of patients and health systems. Semin. Cancer Biol. 2021, 84, 293–301. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Gómez-López, G.; Dopazo, J.; Cigudosa, J.C.; Valencia, A.; Al-Shahrour, F. Precision medicine needs pioneering clinical bioinformaticians. Brief. Bioinform. 2017, 20, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef]
- Singer, J.; Irmisch, A.; Ruscheweyh, H.-J.; Singer, F.; Toussaint, N.C.; Levesque, M.P.; Stekhoven, D.J.; Beerenwinkel, N. Bioinformatics for precision oncology. Brief. Bioinform. 2017, 20, 778–788. [Google Scholar] [CrossRef]
- Gauthier, J.; Vincent, A.T.; Charette, S.J.; Derome, N. A brief history of bioinformatics. Brief. Bioinform. 2018, 20, 1981–1996. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Y.; Zhu, N.; Klau, G.W.; Feng, W. Bioinformatics Methods and Biological Interpretation for Next-Generation Sequencing Data. BioMed Res. Int. 2015, 2015, 690873. [Google Scholar] [CrossRef]
- Nik-Zainal, S. Insights into cancer biology through next-generation sequencing. Clin. Med. 2014, 14, s71–s77. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Patch, A.-M. The Impact of Next Generation Sequencing in Cancer Research. Cancers 2020, 12, 2928. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Melendrez, M.C.; Bishop-Lilly, K.A.; Rutvisuttinunt, W.; Pollett, S.; Talundzic, E.; Morton, L.; Jarman, R.G. Next Generation Sequencing and Bioinformatics Methodologies for Infectious Disease Research and Public Health: Approaches, Applications, and Considerations for Development of Laboratory Capacity. J. Infect. Dis. 2019, 221, S292–S307. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef]
- Beck, T.F.; Mullikin, J.C.; NISC Comparative Sequencing Program. Systematic Evaluation of Sanger Validation of Next-Generation Sequencing Variants. Clin. Chem. 2016, 62, 647–654. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Kumar, A.E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Keshavan, A.; Poline, J.-B. From the Wet Lab to the Web Lab: A Paradigm Shift in Brain Imaging Research. Front. Neuroinform. 2019, 13, 3. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-Generation Sequencing Platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Marra, M.; Friedman, J. Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.Y.; Jones, M.; Jones, S. Whole-Genome Sequencing in Cancer. Cold Spring Harb. Perspect. Med. 2018, 9, a034579. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Balloux, F.; Brynildsrud, O.B.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef]
- Ghazani, A.A.; Oliver, N.M.; Pierre, J.P.S.; Garofalo, A.; Rainville, I.R.; Hiller, E.; Treacy, D.J.; Rojas-Rudilla, V.; Wood, S.; Bair, E.; et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet. Med. 2017, 19, 787–795. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Ulintz, P.J.; Wu, W.; Gates, C.M. Bioinformatics Analysis of Whole Exome Sequencing Data. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1881, pp. 277–318. [Google Scholar] [CrossRef]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-based enrichment strategies for targeted sequencing. Biotechnol. Adv. 2020, 46, 107672. [Google Scholar] [CrossRef]
- Roca, I.; González-Castro, L.; Fernandez-Lopez, H.; Couce, M.L.; Fernández-Marmiesse, A. Free-access copy-number variant detection tools for targeted next-generation sequencing data. Mutat. Res. Mutat. Res. 2019, 779, 114–125. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Bruijns, B.; Tiggelaar, R.M.; Gardeniers, J. Massively parallel sequencing techniques for forensics: A review. Electrophoresis 2018, 39, 2642–2654. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Kramer, M.; Parla, J.; Goes, F.S.; Potash, J.B.; McCombie, W.R.; Zandi, P.P. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum. Genom. 2014, 8, 14. [Google Scholar] [CrossRef]
- Santani, A.; Murrell, J.; Funke, B.; Yu, Z.; Hegde, M.; Mao, R.; Ferreira-Gonzalez, A.; Voelkerding, K.V.; Weck, K.E. Development and Validation of Targeted Next-Generation Sequencing Panels for Detection of Germline Variants in Inherited Diseases. Arch. Pathol. Lab. Med. 2017, 141, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, H.; Banerjee, S.; Li, Y.; Zhou, J.; Yang, Q.; Tan, X.; Han, P.; Fu, Q.; Cui, X.; et al. A comprehensive assessment of Next-Generation Sequencing variants validation using a secondary technology. Mol. Genet. Genom. Med. 2019, 7, e00748. [Google Scholar] [CrossRef]
- Ilyas, M. Next-Generation Sequencing in Diagnostic Pathology. Pathobiology 2017, 84, 292–305. [Google Scholar] [CrossRef]
- Rossing, M.; Sørensen, C.S.; Ejlertsen, B.; Nielsen, F.C. Whole genome sequencing of breast cancer. APMIS 2019, 127, 303–315. [Google Scholar] [CrossRef]
- Garagnani, P.; Marquis, J.; Delledonne, M.; Pirazzini, C.; Marasco, E.; Kwiatkowska, K.M.; Iannuzzi, V.; Bacalini, M.G.; Valsesia, A.; Carayol, J.; et al. Whole-genome sequencing analysis of semi-supercentenarians. eLife 2021, 10, e57849. [Google Scholar] [CrossRef]
- Yoshinaga, Y.; Daum, C.; He, G.; O’Malley, R. Genome Sequencing. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1775, pp. 37–52. [Google Scholar] [CrossRef]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef]
- Aly, S.M.; Sabri, D.M. Next generation sequencing (NGS): A golden tool in forensic toolkit. Arch. Forensic Med. Criminol. 2015, 4, 260–271. [Google Scholar] [CrossRef]
- Verma, M.; Kulshrestha, S.; Puri, A. Genome Sequencing. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1525, pp. 3–33. [Google Scholar] [CrossRef]
- Horner, D.S.; Pavesi, G.; Castrignano, T.; De Meo, P.D.; Liuni, S.; Sammeth, M.; Picardi, E.; Pesole, G. Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief. Bioinform. 2009, 11, 181–197. [Google Scholar] [CrossRef]
- Wang, M.D. In the Spotlight: Bioinformatics. IEEE Rev. Biomed. Eng. 2012, 6, 3–8. [Google Scholar] [CrossRef][Green Version]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- SoRelle, J.A.; Wachsmann, M.; Cantarel, B.L. Assembling and Validating Bioinformatic Pipelines for Next-Generation Sequencing Clinical Assays. Arch. Pathol. Lab. Med. 2020, 144, 1118–1130. [Google Scholar] [CrossRef]
- Gullapalli, R.R. Evaluation of Commercial Next-Generation Sequencing Bioinformatics Software Solutions. J. Mol. Diagn. 2019, 22, 147–158. [Google Scholar] [CrossRef]
- Schwarz, U.I.; Gulilat, M.; Kim, R.B. The Role of Next-Generation Sequencing in Pharmacogenetics and Pharmacogenomics. Cold Spring Harb. Perspect. Med. 2019, 9, a033027. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Van der Auwera, G.A. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Krusche, P.; Trigg, L.; Boutros, P.C.; Mason, C.E.; De La Vega, F.M.; Moore, B.L.; Gonzalez-Porta, M.; Eberle, M.A.; Tezak, Z.; Lababidi, S.; et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat. Biotechnol. 2019, 37, 555–560. [Google Scholar] [CrossRef]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 91. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Lieven, C.; Lieven, C.; Beber, M.E.; Beber, M.E.; Olivier, B.G.; Olivier, B.G.; Bergmann, F.T.; Bergmann, F.T.; Ataman, M.; Ataman, M.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. Available online: https://arxiv.org/abs/1207.3907 (accessed on 1 January 2016).
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Eisfeldt, J.; Vezzi, F.; Olason, P.; Nilsson, D.; Lindstrand, A. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Res 2017, 6, 664. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2015, 12, e1004873. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017. [Google Scholar] [CrossRef]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. BioRxiv 2019. [Google Scholar] [CrossRef]
- Jia, P.; Yang, X.; Guo, L.; Liu, B.; Lin, J.; Liang, H.; Sun, J.; Zhang, C.; Ye, K. MSIsensor-pro: Fast, Accurate, and Matched-normal-sample-free Detection of Microsatellite Instability. Genom. Proteom. Bioinform. 2020, 18, 65–71. [Google Scholar] [CrossRef]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; De Ståhl, T.D.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Research 2020, 9, 63. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef]
- OncoKB: A Precision Oncology Knowledge Base. 2017. Available online: http://oncokb.org (accessed on 12 July 2022).
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion 2018, 50, 71–91. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Zhou, W.; Kang, L.; Duan, H.; Qiao, S.; Tao, L.; Chen, Z.; Huang, Y. A virtual sequencer reveals the dephasing patterns in error-correction code DNA sequencing. Natl. Sci. Rev. 2020, 8, nwaa227. [Google Scholar] [CrossRef]
- Mazlan, A.U.; Sahabudin, N.A.; Remli, M.A.; Ismail, N.S.N.; Mohamad, M.S.; Nies, H.W.; Warif, N.B.A. A Review on Recent Progress in Machine Learning and Deep Learning Methods for Cancer Classification on Gene Expression Data. Processes 2021, 9, 1466. [Google Scholar] [CrossRef]
- Luo, R.; Sedlazeck, F.J.; Lam, T.-W.; Schatz, M.C. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef]
- Sahraeian, S.M.E.; Liu, R.; Lau, B.; Podesta, K.; Mohiyuddin, M.; Lam, H.Y.K. Deep convolutional neural networks for accurate somatic mutation detection. Nat. Commun. 2019, 10, 1041. [Google Scholar] [CrossRef]
- Cai, L.; Wu, Y.; Gao, J. DeepSV: Accurate calling of genomic deletions from high-throughput sequencing data using deep convolutional neural network. BMC Bioinform. 2019, 20, 665. [Google Scholar] [CrossRef]
- Friedman, S.; Gauthier, L.; Farjoun, Y.; Banks, E. Lean and deep models for more accurate filtering of SNP and INDEL variant calls. Bioinformatics 2019, 36, 2060–2067. [Google Scholar] [CrossRef]
- Kohestani, H.; Giuliani, A. Organization principles of biological networks: An explorative study. Biosystems 2016, 141, 31–39. [Google Scholar] [CrossRef]
- Norori, N.; Hu, Q.; Aellen, F.M.; Faraci, F.D.; Tzovara, A. Addressing bias in big data and AI for health care: A call for open science. Gene Expr. Patterns 2021, 2, 100347. [Google Scholar] [CrossRef]
- Weissler, E.H.; Naumann, T.; Andersson, T.; Ranganath, R.; Elemento, O.; Luo, Y.; Freitag, D.F.; Benoit, J.; Hughes, M.C.; Khan, F.; et al. The role of machine learning in clinical research: Transforming the future of evidence generation. Trials 2021, 22, 537. [Google Scholar] [CrossRef]
- Beaulieu-Jones, B.K.; Yuan, W.; Brat, G.A.; Beam, A.L.; Weber, G.; Ruffin, M.; Kohane, I.S. Machine learning for patient risk stratification: Standing on, or looking over, the shoulders of clinicians? NPJ Digit. Med. 2021, 4, 62. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef]
- Mallard, C.; Johnston, M.J.; Bobyn, A.; Nikolic, A.; Argiropoulos, B.; Chan, J.A.; Guilcher, G.M.; Gallo, M. Hi-C detects genomic structural variants in peripheral blood of pediatric leukemia patients. Mol. Case Stud. 2022, 8, a006157. [Google Scholar] [CrossRef]
- Shigaki, D.; Adato, O.; Adhikari, A.N.; Dong, S.; Hawkins-Hooker, A.; Inoue, F.; Juven-Gershon, T.; Kenlay, H.; Martin, B.; Patra, A.; et al. Integration of multiple epigenomic marks improves prediction of variant impact in saturation mutagenesis reporter assay. Hum. Mutat. 2019, 40, 1280–1291. [Google Scholar] [CrossRef]
- Tan, J.; Doing, G.; Lewis, K.A.; Price, C.E.; Chen, K.M.; Cady, K.C.; Perchuk, B.; Laub, M.T.; Hogan, D.A.; Greene, C.S. Unsupervised Extraction of Stable Expression Signatures from Public Compendia with an Ensemble of Neural Networks. Cell Syst. 2017, 5, 63–71.e6. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. Ten-sorflow: Large-scale machine learning on heterogeneous distributed systems. arXiv 2004, arXiv:1603.04467. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. 2019. Available online: http://arxiv.org/abs/1912.01703 (accessed on 12 July 2022).
- Collobert, R.; Kavukcuoglu, K.; Farabet, C. Torch7: A Matlab-Like Environment for Machine Learning. Available online: http://numpy.scipy.org (accessed on 12 July 2022).
- Chartrand, G.; Zhang, P. Introduction to Graphs. In Chromatic Graph Theory; Chapman and Hall/CRC: New York, NY, USA, 2019; pp. 27–52. [Google Scholar] [CrossRef]
- Ghosh, S.; Mukherjee, S.; Sengupta, N.; Roy, A.; Dey, D.; Chakraborty, S.; Chattopadhyay, D.; Banerjee, A.; Basu, A. Network analysis reveals common host protein/s modulating pathogenesis of neurotropic viruses. Sci. Rep. 2016, 6, 32593. [Google Scholar] [CrossRef]
- Huang, C.-H.; Zaenudin, E.; Tsai, J.J.; Kurubanjerdjit, N.; Dessie, E.Y.; Ng, K.-L. Dissecting molecular network structures using a network subgraph approach. PeerJ 2020, 8, e9556. [Google Scholar] [CrossRef]
- Huang, C.-H.; Zaenudin, E.; Tsai, J.J.; Kurubanjerdjit, N.; Ng, K.-L. Network subgraph-based approach for analyzing and comparing molecular networks. PeerJ 2022, 10, e13137. [Google Scholar] [CrossRef]
- Torshizi, A.D.; Petzold, L.R. Graph-based semi-supervised learning with genomic data integration using condition-responsive genes applied to phenotype classification. J. Am. Med. Inform. Assoc. 2017, 25, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Mentzelopoulos, A.; Karanasiou, I.; Papathanasiou, M.; Kelekis, N.; Kouloulias, V.; Matsopoulos, G.K. A Comparative Analysis of White Matter Structural Networks on SCLC Patients After Chemotherapy. Brain Topogr. 2022, 35, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G. The Igraph Software Package for Complex Network Research. Available online: https://www.researchgate.net/publication/221995787 (accessed on 12 July 2022).
- Mueller, L.A.J.; Kugler, K.G.; Dander, A.; Graber, A.; Dehmer, M. QuACN: An R package for analyzing complex biological networks quantitatively. Bioinformatics 2010, 27, 140–141. [Google Scholar] [CrossRef] [PubMed]
- Handcock, M.S.; Hunter, D.R.; Butts, C.T.; Goodreau, S.M.; Morris, M. Analysis and Simulation of Network Data. Available online: http://CRAN.R-project.org/ (accessed on 12 July 2022).
- Tripathi, S.; Dehmer, M.; Emmert-Streib, F. NetBioV: An R package for visualizing large network data in biology and medicine. Bioinformatics 2014, 30, 2834–2836. [Google Scholar] [CrossRef]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Bollen, K.A. Structural Equations with Latent Variables; Wiley: Hoboken, NJ, USA, 1989. [Google Scholar]
- Dellino, G.I.; Palluzzi, F.; Chiariello, A.M.; Piccioni, R.; Bianco, S.; Furia, L.; De Conti, G.; Bouwman, B.A.M.; Melloni, G.; Guido, D.; et al. Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nat. Genet. 2019, 51, 1011–1023. [Google Scholar] [CrossRef]
- Saranya, A.; Venkatesan, S. A Model Based Approach on Gene Expression Profiling of Colorectal Cancer and Normal Mucosa Using Logistic Regression, Artificial Neural Network and Structural Equation Modelling. Turk. J. Comput. Math. Educ. 2021, 12, 2585–2593. [Google Scholar]
- Pepe, D.; Do, J.H. Estimation of dysregulated pathway regions in MPP+ treated human neuroblastoma SH-EP cells with structural equation model. BioChip J. 2015, 9, 131–138. [Google Scholar] [CrossRef]
- Mogaka, J.J.O.; Chimbari, M.J. The mediating effects of public genomic knowledge in precision medicine implementation: A structural equation model approach. PLoS ONE 2020, 15, e0240585. [Google Scholar] [CrossRef]
- Rosseel, Y. Journal of Statistical Software lavaan: An R Package for Structural Equation Modeling. 2012. Available online: http://www.jstatsoft.org/ (accessed on 12 July 2022).
- Palluzzi, F.; Grassi, M. SEMgraph: An R Package for Causal Network Analysis of High-Throughput Data with Structural Equation Models. 2021. Available online: http://arxiv.org/abs/2103.08332 (accessed on 12 July 2022).
- Verhulst, B.; Maes, H.H.; Neale, M.C. GW-SEM: A Statistical Package to Conduct Genome-Wide Structural Equation Modeling. Behav. Genet. 2017, 47, 345–359. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Niraparib: First Global Approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Ethier, J.-L.; Lheureux, S.; Oza, A.M. The role of niraparib for the treatment of ovarian cancer. Future Oncol. 2018, 14, 2565–2577. [Google Scholar] [CrossRef]
- Ison, G.; Howie, L.J.; Amiri-Kordestani, L.; Zhang, L.; Tang, S.; Sridhara, R.; Pierre, V.; Charlab, R.; Ramamoorthy, A.; Song, P.; et al. FDA Approval Summary: Niraparib for the Maintenance Treatment of Patients with Recurrent Ovarian Cancer in Response to Platinum-Based Chemotherapy. Clin. Cancer Res. 2018, 24, 4066–4071. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. JNCI J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Kang, H.G.; Hwangbo, H.; Kim, M.J.; Kim, S.; Lee, E.J.; Park, M.J.; Kim, J.-W.; Kim, B.-G.; Cho, E.-H.; Chang, S.; et al. Aberrant Transcript Usage Is Associated with Homologous Recombination Deficiency and Predicts Therapeutic Response. Cancer Res. 2022, 82, 142–154. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef]
- Foote, J.R.; Lopez-Acevedo, M.; Buchanan, A.H.; Secord, A.A.; Lee, P.S.; Fountain, C.; Myers, E.R.; Cohn, D.E.; Reed, S.D.; Havrilesky, L.J. Cost Comparison of Genetic Testing Strategies in Women with Epithelial Ovarian Cancer. J. Oncol. Pract. 2017, 13, e120–e129. [Google Scholar] [CrossRef]
- McLaughlin, L.J.; Stojanovic, L.; Kogan, A.A.; Rutherford, J.L.; Choi, E.Y.; Yen, R.-W.C.; Xia, L.; Zou, Y.; Lapidus, R.G.; Baylin, S.B.; et al. Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc. Natl. Acad. Sci. USA 2020, 117, 17785–17795. [Google Scholar] [CrossRef] [PubMed]
- Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Siemanowski, J. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Pers. Med. 2021, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Shirts, B.H.; Casadei, S.; Jacobson, A.L.; Lee, M.K.; Gulsuner, S.; Bennett, R.L.; Miller, M.; Hall, S.A.; Hampel, H.; Hisama, F.M.; et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet. Med. 2016, 18, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef]
- Kurian, A.W.; Kingham, K.E.; Ford, J.M. Next-generation sequencing for hereditary breast and gynecologic cancer risk assessment. Curr. Opin. Obstet. Gynecol. 2015, 27, 23–33. [Google Scholar] [CrossRef]
- Telli, M.L.; Stover, D.G.; Loi, S.; Aparicio, S.; Carey, L.A.; Domchek, S.M.; Newman, L.; Sledge, G.W.; Winer, E.P. Homologous recombination deficiency and host anti-tumor immunity in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 171, 21–31. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef]
- De Luca, X.M.; Newell, F.; Kazakoff, S.H.; Hartel, G.; Reed, A.E.M.; Holmes, O.; Xu, Q.; Wood, S.; Leonard, C.; Pearson, J.V.; et al. Using whole-genome sequencing data to derive the homologous recombination deficiency scores. NPJ Breast Cancer 2020, 6, 33. [Google Scholar] [CrossRef]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Tumiati, M.; Hietanen, S.; Hynninen, J.; Pietilä, E.; Färkkilä, A.; Kaipio, K.; Roering, P.; Huhtinen, K.; Alkodsi, A.; Li, Y.; et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res. 2018, 24, 4482–4493. [Google Scholar] [CrossRef] [PubMed]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- MacIntyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy-number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reuterswärd, C.; Häkkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Borcsok, J.; Prosz, A.; Cornelius, N.; Kjeldsen, M.K.; Mirza, M.R.; Szallasi, Z. Comparative Assessment of Diagnostic Homologous Recombination Deficiency–Associated Mutational Signatures in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 5681–5687. [Google Scholar] [CrossRef]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e9. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, A.J.; Lee, J.-K.; Park, J.; Kwon, Y.; Park, S.; Chun, H.; Ju, Y.S.; Hong, D. Mutalisk: A web-based somatic MUTation AnaLyIS toolKit for genomic, transcriptional and epigenomic signatures. Nucleic Acids Res. 2018, 46, W102–W108. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.-K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Matondo, A.; Jo, Y.H.; Shahid, M.; Choi, T.G.; Nguyen, M.N.; Nguyen, N.N.Y.; Akter, S.; Kang, I.; Ha, J.; Maeng, C.H.; et al. The Prognostic 97 Chemoresponse Gene Signature in Ovarian Cancer. Sci. Rep. 2017, 7, 9689. [Google Scholar] [CrossRef]
- Leibowitz, B.D.; Dougherty, B.V.; Bell, J.S.K.; Kapilivsky, J.; Michuda, J.; Sedgewick, A.J.; Munson, W.A.; Chandra, T.A.; Dry, J.R.; Beaubier, N.; et al. Validation of genomic and transcriptomic models of homologous recombination deficiency in a real-world pan-cancer cohort. BMC Cancer 2022, 22, 587. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Ai, L.; Wang, Y.; Liu, K.; Chen, B.; Chen, T.; Zhuang, S.; Xu, H.; Zou, M.; et al. Discovering a qualitative transcriptional signature of homologous recombination defectiveness for prostate cancer. iScience 2021, 24, 103135. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shao, M.; Meng, P.; Wang, C.; Li, Q.; Cai, Y.; Song, C.; Wang, X.; Shi, T. GSA: An independent development algorithm for calling copy number and detecting homologous recombination deficiency (HRD) from target capture sequencing. BMC Bioinform. 2021, 22, 562. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Bosquet, J.; Newtson, A.M.; Chung, R.K.; Thiel, K.W.; Ginader, T.; Goodheart, M.J.; Leslie, K.K.; Smith, B.J. Prediction of chemo-response in serous ovarian cancer. Mol. Cancer 2016, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Lai, C.-H.; Wang, T.-H.; Jung, S.-M.; Lee, Y.-S.; Chang, W.-Y.; Yang, L.-Y.; Ku, F.-C.; Huang, H.-J.; Chao, A.-S.; et al. Genomic scar signatures associated with homologous recombination deficiency predict adverse clinical outcomes in patients with ovarian clear cell carcinoma. Klin. Wochenschr. 2018, 96, 527–536. [Google Scholar] [CrossRef]
- Peng, G.; Lin, C.C.-J.; Mo, W.; Dai, H.; Park, Y.-Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Chakravarty, D.; Johnson, A.; Sklar, J.; Lindeman, N.I.; Moore, K.; Ganesan, S.; Lovly, C.M.; Perlmutter, J.; Gray, S.W.; Hwang, J.; et al. Somatic Genomic Testing in Patients with Metastatic or Advanced Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2022, 40, 1231–1258. [Google Scholar] [CrossRef]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Liu, Y.L.; Selenica, P.; Zhou, Q.; Iasonos, A.; Callahan, M.; Feit, N.Z.; Boland, J.; Vazquez-Garcia, I.; Mandelker, D.; Zehir, A.; et al. BRCA Mutations, Homologous DNA Repair Deficiency, Tumor Mutational Burden, and Response to Immune Checkpoint Inhibition in Recurrent Ovarian Cancer. JCO Precis. Oncol. 2020, 4, 665–679. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tool | Sample Type | Variant Type | Method | Ref |
|---|---|---|---|---|
| Manta | G, S | SV, indels | Graph-based breakend analysis | [57] |
| TIDDIT | G, S | SV | Coverage-based genome scan | [58] |
| Cnvkit | G, S | CNV | Coverage-based genome scan | [59] |
| Freebayes | G, S | SNV, indels | Haplotype-based Bayes theorem | [60] |
| Strelka2 | G, S | SNV, indels | Haplotype-based mixture modeling | [61] |
| DeepVariant | G | SNV, indels | Pileup image CNN classification | [62] |
| HaplotypeCaller | G | SNV, indels | Haplotype re-assembly, likelihood | [63] |
| Mpileup | G | SNV, indels | Local re-alignment, likelihood | [55] |
| Mutect2 | S | SNV, indels | GATK + read-to-haplotype alignment | [64] |
| Ascat | S | CNV | Signal intensity and allele frequency | [65] |
| Control-FREEC | S | CNV | LASSO-based genome segmentation | [66] |
| MSIsensor-pro | S | MSI | Multinomial distribution | [67] |
| Tools | Applications | Variants Type | References |
|---|---|---|---|
| HRDetect | WGS | indels, snv, sv and CNV | [146] |
| Mutalisk | WGS, WES and TS | Mutational signatures | [147] |
| SigMA | WGS, WES and TS | SNV | [148] |
| CHORD | WGS | SNV, indels | [153,154] |
| PathAI | WGS, WES and TS | Indels, snv | https://www.pathai.com (accessed on 12 July 2022) |
| GSA | WGS, WES and TS | CNV | [156] |
| AcornHRD | WGS, WES and TS | Indels, CNV and snv | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dotolo, S.; Esposito Abate, R.; Roma, C.; Guido, D.; Preziosi, A.; Tropea, B.; Palluzzi, F.; Giacò, L.; Normanno, N. Bioinformatics: From NGS Data to Biological Complexity in Variant Detection and Oncological Clinical Practice. Biomedicines 2022, 10, 2074. https://doi.org/10.3390/biomedicines10092074
Dotolo S, Esposito Abate R, Roma C, Guido D, Preziosi A, Tropea B, Palluzzi F, Giacò L, Normanno N. Bioinformatics: From NGS Data to Biological Complexity in Variant Detection and Oncological Clinical Practice. Biomedicines. 2022; 10(9):2074. https://doi.org/10.3390/biomedicines10092074
Chicago/Turabian StyleDotolo, Serena, Riziero Esposito Abate, Cristin Roma, Davide Guido, Alessia Preziosi, Beatrice Tropea, Fernando Palluzzi, Luciano Giacò, and Nicola Normanno. 2022. "Bioinformatics: From NGS Data to Biological Complexity in Variant Detection and Oncological Clinical Practice" Biomedicines 10, no. 9: 2074. https://doi.org/10.3390/biomedicines10092074
APA StyleDotolo, S., Esposito Abate, R., Roma, C., Guido, D., Preziosi, A., Tropea, B., Palluzzi, F., Giacò, L., & Normanno, N. (2022). Bioinformatics: From NGS Data to Biological Complexity in Variant Detection and Oncological Clinical Practice. Biomedicines, 10(9), 2074. https://doi.org/10.3390/biomedicines10092074