
The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer

 , ,
, , 
Abstract
:1. Introduction
2. The Development of Prostate Cancer Immunotherapy
2.1. Sipuleucel-T
2.2. Adoptive Immune Cell Immunotherapy
2.3. Immune Checkpoint Inhibitors
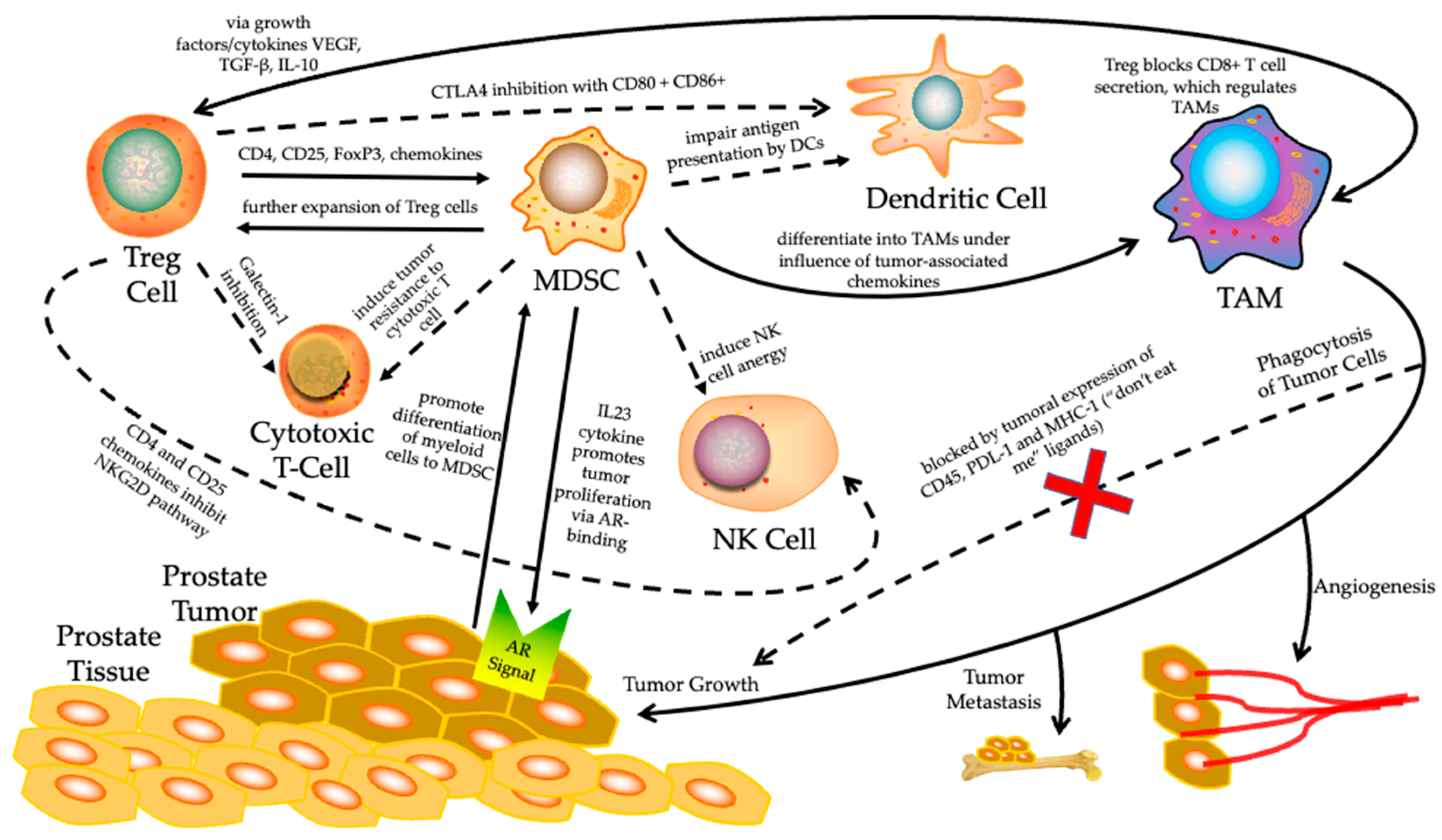
3. Role of TME in Therapy-Resistant Prostate Cancer Immune Evasion
3.1. Regulation of Tumor Immune Microenvironment (TIME)
3.2. The Role of MDSC in Prostate Cancer Immune Evasion
3.3. The Treg Cells in Prostate Cancer Immune Evasion
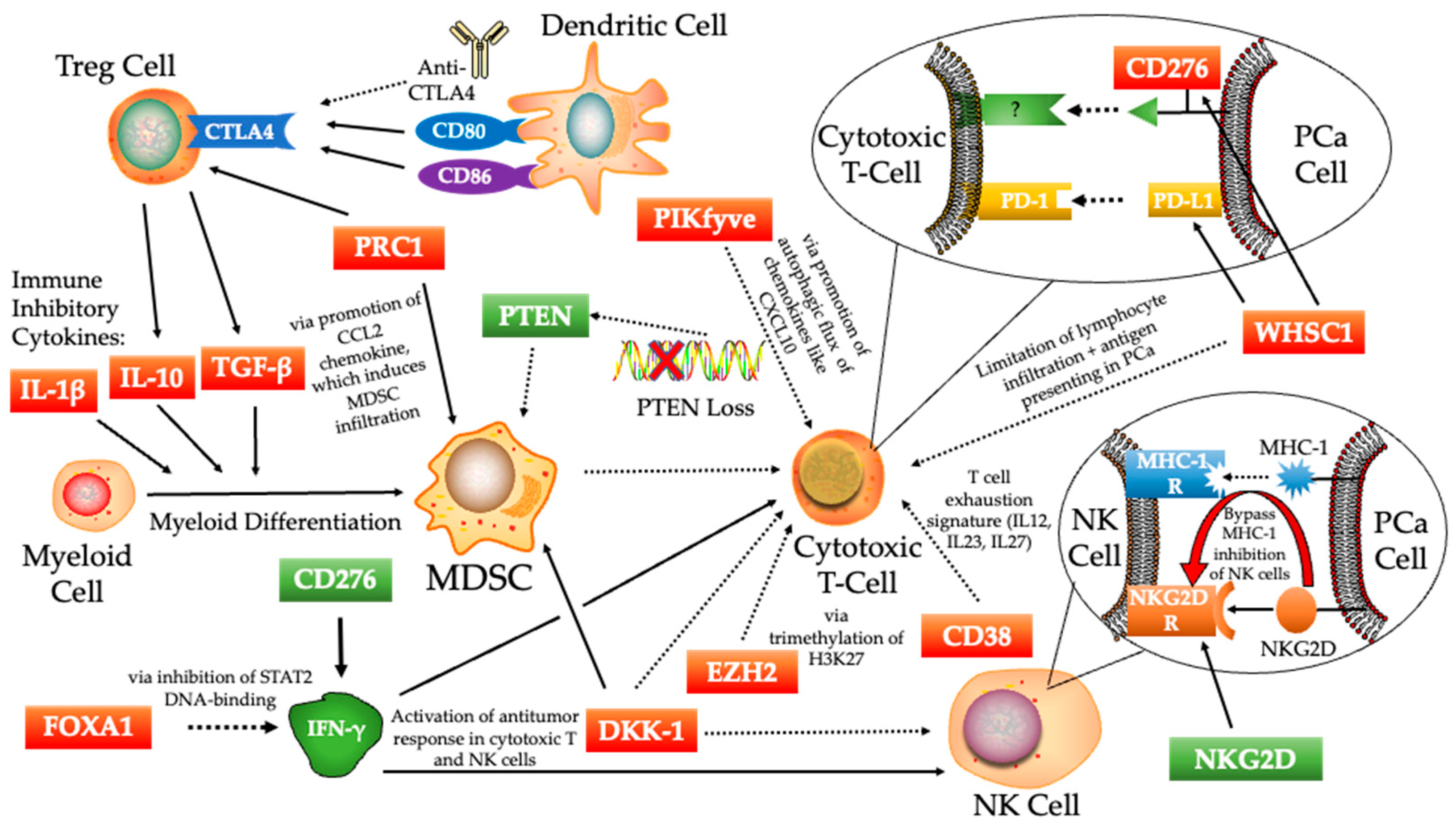
4. The Molecular Mechanisms of Immunosuppressive Signaling Activation in Prostate Cancer
4.1. CD276 (B7-H3)
4.2. PTEN
4.3. FOXA1
4.4. EZH2
4.5. DKK-1
4.6. WHSC1
4.7. NKG2D
4.8. CD38
4.9. PRC1
4.10. PIKfyve
5. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II-2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef]
- Achard, V.; Putora, P.M.; Omlin, A.; Zilli, T.; Fischer, S. Metastatic Prostate Cancer: Treatment Options. Oncology 2022, 100, 48–59. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Lallous, N.; Snow, O.; Sanchez, C.; Parra Nuñez, A.K.; Sun, B.; Hussain, A.; Lee, J.; Morin, H.; Leblanc, E.; Gleave, M.E.; et al. Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients. Cancers 2021, 13, 2939. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Uemura, M.; Tamura, K.; Chung, S.; Honma, S.; Okuyama, A.; Nakamura, Y.; Nakagawa, H. Novel 5 alpha-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Sci. 2008, 99, 81–86. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/beta-Catenin Pathway Overcomes Resistance to Enzalutamide in Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Shields, M.D.; Marin-Acevedo, J.A.; Pellini, B. Immunotherapy for Advanced Non-Small Cell Lung Cancer: A Decade of Progress. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, 1–23. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Guo, C.; Gurel, B.; De Marzo, A.M.; Sfanos, K.S.; Mani, R.S.; Gil, J.; Drake, C.G.; Alimonti, A. Prostate carcinogenesis: Inflammatory storms. Nat. Rev. Cancer 2020, 20, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Krueger, T.E.; Thorek, D.L.J.; Meeker, A.K.; Isaacs, J.T.; Brennen, W.N. Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate 2019, 79, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Nava Rodrigues, D.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 4441–4453. [Google Scholar] [CrossRef] [Green Version]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The Tumor Immune Contexture of Prostate Cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.B.; Guedes, L.B.; Lu, J.; Maldonado, L.; Reitz, L.; Barber, J.R.; De Marzo, A.M.; Tosoian, J.J.; Tomlins, S.A.; Schaeffer, E.M.; et al. Association of tumor-infiltrating T-cell density with molecular subtype, racial ancestry and clinical outcomes in prostate cancer. Mod. Pathol. 2018, 31, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Bernstein, G.T.; Corman, J.M.; Glode, L.M.; Hall, S.J.; Poll, W.L.; Schellhammer, P.F.; Jones, L.A.; Xu, Y.; Kylstra, J.W.; et al. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4558–4567. [Google Scholar] [CrossRef] [Green Version]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Hafron, J.M.; Wilfehrt, H.M.; Ferro, C.; Harmon, M.; Flanders, S.C.; McKay, R.R. Real-World Effectiveness of Sipuleucel-T on Overall Survival in Men with Advanced Prostate Cancer Treated with Androgen Receptor-Targeting Agents. Adv. Ther. 2022, 39, 2515–2532. [Google Scholar] [CrossRef]
- Dorff, T.; Hirasawa, Y.; Acoba, J.; Pagano, I.; Tamura, D.; Pal, S.; Zhang, M.; Waitz, R.; Dhal, A.; Haynes, W.; et al. Phase Ib study of patients with metastatic castrate-resistant prostate cancer treated with different sequencing regimens of atezolizumab and sipuleucel-T. J. Immunother. Cancer 2021, 9, e002931. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.H.; Fu, W.; Wang, H.; Park, J.C.; DeWeese, T.L.; Tran, P.T.; Song, D.Y.; King, S.; Afful, M.; Hurrelbrink, J.; et al. Randomized Phase II Trial of Sipuleucel-T with or without Radium-223 in Men with Bone-metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Cappuccini, F.; Bryant, R.; Pollock, E.; Carter, L.; Verrill, C.; Hollidge, J.; Poulton, I.; Baker, M.; Mitton, C.; Baines, A.; et al. Safety and immunogenicity of novel 5T4 viral vectored vaccination regimens in early stage prostate cancer: A phase I clinical trial. J. Immunother. Cancer 2020, 8, e000928. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Arai, G.; Egawa, S.; Ohyama, C.; Naito, S.; Matsumoto, K.; Uemura, H.; Nakagawa, M.; Nasu, Y.; Eto, M.; et al. Mixed 20-peptide cancer vaccine in combination with docetaxel and dexamethasone for castration-resistant prostate cancer: A randomized phase II trial. Cancer Immunol. Immunother. 2020, 69, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef]
- Slovin, S.F.; Dorff, T.B.; Falchook, G.S.; Wei, X.X.; Gao, X.; McKay, R.R.; Oh, D.Y.; Wibmer, A.G.; Spear, M.A.; McCaigue, J.; et al. Phase 1 study of P-PSMA-101 CAR-T cells in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 98. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Loriot, Y.; Shaffer, D.R.; Braiteh, F.; Powderly, J.; Harshman, L.C.; Conkling, P.; Delord, J.P.; Gordon, M.; Kim, J.W.; et al. Safety and Clinical Activity of Atezolizumab in Patients with Metastatic Castration-Resistant Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2021, 27, 3360–3369. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E., 3rd; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: A randomized phase 3 trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Shenderov, E.; Boudadi, K.; Fu, W.; Wang, H.; Sullivan, R.; Jordan, A.; Dowling, D.; Harb, R.; Schonhoft, J.; Jendrisak, A.; et al. Nivolumab plus ipilimumab, with or without enzalutamide, in AR-V7-expressing metastatic castration-resistant prostate cancer: A phase-2 nonrandomized clinical trial. Prostate 2021, 81, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Flechon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e483. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Carabasi, M.H.; McKean, M.; Stein, M.N.; Schweizer, M.T.; Luke, J.J.; Narayan, V.; Pachynski, R.K.; Parikh, R.A.; Zhang, J.; Fountaine, T.J.; et al. PSMA targeted armored chimeric antigen receptor (CAR) T-cells in patients with advanced mCRPC: A phase I experience. J. Clin. Oncol. 2021, 39, 2534. [Google Scholar] [CrossRef]
- Dorff, T.B.; Blanchard, S.; Martirosyan, H.; Adkins, L.; Dhapola, G.; Moriarty, A.; Wagner, J.R.; Chaudhry, A.; D’Apuzzo, M.; Kuhn, P.; et al. Phase 1 study of PSCA-targeted chimeric antigen receptor (CAR) T cell therapy for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 91. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, Y.; Shi, C. Targeting Natural Killer Cells for Tumor Immunotherapy. Front. Immunol. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Drake, C.G.; Beer, T.M.; Kwon, E.D.; Scher, H.I.; Gerritsen, W.R.; Bossi, A.; den Eertwegh, A.J.M.v.; Krainer, M.; Houede, N.; et al. Final Analysis of the Ipilimumab Versus Placebo Following Radiotherapy Phase III Trial in Postdocetaxel Metastatic Castration-resistant Prostate Cancer Identifies an Excess of Long-term Survivors. Eur. Urol. 2020, 78, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Polesso, F.; Wang, C.; Sehrawat, A.; Hawkins, R.M.; Murray, S.E.; Thomas, G.V.; Caruso, B.; Thompson, R.F.; Wood, M.A.; et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 2022, 606, 791–796. [Google Scholar] [CrossRef]
- Fenner, A. Patient selection is key in IMbassador250. Nat. Rev. Urol. 2022, 19, 131. [Google Scholar] [CrossRef]
- Tarrar, T.A.; Anwar, M.Y.; Ali, M.A.; Saeed, M.; Rehman, S.; Bajwa, S.F.; Ayub, T.; Javid, H.; Ali, R.; Irshad, A.; et al. Current Status of Monoclonal Antibodies-Based Therapies in Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis of Clinical Trials. Cureus 2022, 14, e22942. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Bryant, G.; Wang, L.; Mulholland, D.J. Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer. Int. J. Mol. Sci. 2017, 18, 1542. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haanen, J.B.; Baars, A.; Gomez, R.; Weder, P.; Smits, M.; de Gruijl, T.D.; von Blomberg, B.M.; Bloemena, E.; Scheper, R.J.; van Ham, S.M.; et al. Melanoma-specific tumor-infiltrating lymphocytes but not circulating melanoma-specific T cells may predict survival in resected advanced-stage melanoma patients. Cancer Immunol. Immunother. CII 2006, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Strasner, A.; Karin, M. Immune Infiltration and Prostate Cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Tourinho-Barbosa, R.R.; de la Rosette, J.; Sanchez-Salas, R. Prostate cancer multifocality, the index lesion, and the microenvironment. Curr. Opin. Urol. 2018, 28, 499–505. [Google Scholar] [CrossRef]
- Adekoya, T.O.; Richardson, R.M. Cytokines and Chemokines as Mediators of Prostate Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 4449. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Guo, W.; Liang, X. Phenotypes, accumulation, and functions of myeloid-derived suppressor cells and associated treatment strategies in cancer patients. Hum. Immunol. 2014, 75, 1128–1137. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Greten, T.F.; Manns, M.P.; Korangy, F. Myeloid derived suppressor cells in human diseases. Int. Immunopharmacol. 2011, 11, 802–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; McGuire, T.R.; Britton, H.C.; Schwarz, J.K.; Loberiza, F.R., Jr.; Meza, J.L.; Talmadge, J.E. Lenalidomide and cyclophosphamide immunoregulation in patients with metastatic, castration-resistant prostate cancer. Clin. Exp. Metastasis 2015, 32, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Køllgaard, T.; Kongsted, P.; Sengeløv, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. CII 2014, 63, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 10094–10099. [Google Scholar] [CrossRef] [Green Version]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-Derived Suppressor Cells in Prostate Cancer: Present Knowledge and Future Perspectives. Cells 2021, 11, 20. [Google Scholar] [CrossRef]
- Askenasy, N.; Kaminitz, A.; Yarkoni, S. Mechanisms of T regulatory cell function. Autoimmun. Rev. 2008, 7, 370–375. [Google Scholar] [CrossRef]
- Garín, M.I.; Chu, C.C.; Golshayan, D.; Cernuda-Morollón, E.; Wait, R.; Lechler, R.I. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007, 109, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Peach, R.J.; Bajorath, J.; Brady, W.; Leytze, G.; Greene, J.; Naemura, J.; Linsley, P.S. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J. Exp. Med. 1994, 180, 2049–2058. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Mathur, R.; Farooque, A.; Kaul, V.; Gupta, S.; Dwarakanath, B.S. T-Regulatory Cells In Tumor Progression And Therapy. Cancer Manag. Res. 2019, 11, 10731–10747. [Google Scholar] [CrossRef] [Green Version]
- Karpisheh, V.; Mousavi, S.M.; Naghavi Sheykholeslami, P.; Fathi, M.; Mohammadpour Saray, M.; Aghebati-Maleki, L.; Jafari, R.; Majidi Zolbanin, N.; Jadidi-Niaragh, F. The role of regulatory T cells in the pathogenesis and treatment of prostate cancer. Life Sci. 2021, 284, 119132. [Google Scholar] [CrossRef]
- Klyushnenkova, E.N.; Riabov, V.B.; Kouiavskaia, D.V.; Wietsma, A.; Zhan, M.; Alexander, R.B. Breaking immune tolerance by targeting CD25+ regulatory T cells is essential for the anti-tumor effect of the CTLA-4 blockade in an HLA-DR transgenic mouse model of prostate cancer. Prostate 2014, 74, 1423–1432. [Google Scholar] [CrossRef]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zennami, K.; et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PloS ONE 2012, 7, e30815. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Yonesaka, K.; Haratani, K.; Takamura, S.; Sakai, H.; Kato, R.; Takegawa, N.; Takahama, T.; Tanaka, K.; Hayashi, H.; Takeda, M.; et al. B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2653–2664. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, D.-C.; Zhu, X.-G.; Gan, W.-J.; Li, Z.; Xiong, F.; Zhang, Z.-X.; Zhang, G.-B.; Zhang, X.-G.; Zhao, H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int. J. Mol. Med. 2013, 31, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Suh, W.K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A.; et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, P.; Li, J.; Zhao, J.; Liu, C.; Yang, F.; Yang, D.; Gao, A.; Lin, W.; Ma, X.; et al. B7-H3 in combination with regulatory T cell is associated with tumor progression in primary human non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13987–13995. [Google Scholar] [PubMed]
- Liu, C.; Liu, J.; Wang, J.; Liu, Y.; Zhang, F.; Lin, W.; Gao, A.; Sun, M.; Wang, Y.; Sun, Y. B7-H3 expression in ductal and lobular breast cancer and its association with IL-10. Mol. Med. Rep. 2013, 7, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Kang, F.-B.; Wang, L.; Jia, H.-C.; Li, D.; Li, H.-J.; Zhang, Y.-G.; Sun, D.-X. B7-H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial-to-mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int. 2015, 15, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flem-Karlsen, K.; Tekle, C.; Andersson, Y.; Flatmark, K.; Fodstad, Ø.; Nunes-Xavier, C.E. Immunoregulatory protein B7-H3 promotes growth and decreases sensitivity to therapy in metastatic melanoma cells. Pigment. Cell Melanoma Res. 2017, 30, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Xavier, C.E.; Karlsen, K.F.; Tekle, C.; Pedersen, C.; Øyjord, T.; Hongisto, V.; Nesland, J.M.; Tan, M.; Sahlberg, K.K.; Fodstad, Ø. Decreased expression of B7-H3 reduces the glycolytic capacity and sensitizes breast cancer cells to AKT/mTOR inhibitors. Oncotarget 2016, 7, 6891–6901. [Google Scholar] [CrossRef] [Green Version]
- Amori, G.; Sugawara, E.; Shigematsu, Y.; Akiya, M.; Kunieda, J.; Yuasa, T.; Yamamoto, S.; Yonese, J.; Takeuchi, K.; Inamura, K. Tumor B7-H3 expression in diagnostic biopsy specimens and survival in patients with metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 767–774. [Google Scholar] [CrossRef]
- Guo, C.; Figueiredo, I.; Gurel, B.; Crespo, M.; Rekowski, J.; Carreira, S.; Neeb, A.; Sharp, A.; Fenor de la Maza, M.D.; Rescigno, P.; et al. Abstract LB035: B7-H3 as a therapeutic target in prostate cancer. Cancer Res. 2021, 81, LB035. [Google Scholar] [CrossRef]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar]
- Kulac, I.; Roudier, M.P.; Haffner, M.C. Molecular Pathology of Prostate Cancer. Surg. Pathol. Clin. 2021, 14, 387–401. [Google Scholar] [CrossRef]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; Picanço de Albuquerque, C.G.; Reis, R.B.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 79, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Lentsch, A.B. Progressive dysregulation of transcription factors NF-κB and STAT1 in prostate cancer cells causes proangiogenic production of CXC chemokines. Am. J. Physiol.-Cell Physiol. 2004, 286, C840–C847. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Mendes, A.A.; Lu, J.; Kaur, H.B.; Zheng, S.L.; Xu, J.; Hicks, J.; Weiner, A.B.; Schaeffer, E.M.; Ross, A.E.; Balk, S.P.; et al. Association of B7-H3 expression with racial ancestry, immune cell density, and androgen receptor activation in prostate cancer. Cancer 2022, 128, 2269–2280. [Google Scholar] [CrossRef]
- Conley-LaComb, M.K.; Saliganan, A.; Kandagatla, P.; Chen, Y.Q.; Cher, M.L.; Chinni, S.R. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol. Cancer 2013, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.J.; Neisen, J.; Messenger, J.; Waugh, D.J. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget 2014, 5, 4895–4908. [Google Scholar] [CrossRef] [Green Version]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Zhao, J.C.; Wu, L.; Kim, J.; Yu, J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014, 5, 3972. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Park, S.H.; Zhao, J.C.; Fong, K.W.; Li, S.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.; Abdulkadir, S.A.; et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J. Clin. Investig. 2019, 129, 569–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Fong, K.W.; Kim, J.; Wang, F.; Lu, X.; Lee, Y.; Brea, L.T.; Wadosky, K.; Guo, C.; Abdulkadir, S.A.; et al. Posttranslational regulation of FOXA1 by Polycomb and BUB3/USP7 deubiquitin complex in prostate cancer. Sci. Adv. 2021, 7, eabe2261. [Google Scholar] [CrossRef]
- Brea, L.T.; Wang, X.; Yu, J. Epithelial Transcription Factor FOXA1 Regulates Prostate Cancer Immune Response. J. Endocr. Soc. 2021, 5, A1017. [Google Scholar] [CrossRef]
- He, Y.; Wang, L.; Wei, T.; Xiao, Y.-T.; Sheng, H.; Su, H.; Hollern, D.P.; Zhang, X.; Ma, J.; Wen, S.; et al. FOXA1 overexpression suppresses interferon signaling and immune response in cancer. J. Clin. Investig. 2021, 131, e147025. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Ennishi, D.; Takata, K.; Béguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019, 9, 546–563. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Cañadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA–STING–interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, W.; Xie, L.; Wang, J.; Deng, Y.; Peng, Q.; Zhai, L.; Li, S.; Qin, X. Prognostic significance of dickkopf-1 overexpression in solid tumors: A meta-analysis. Tumour. Biol. 2014, 35, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- Chae, W.J.; Park, J.H.; Henegariu, O.; Yilmaz, S.; Hao, L.; Bothwell, A.L.M. Membrane-bound Dickkopf-1 in Foxp3(+) regulatory T cells suppresses T-cell-mediated autoimmune colitis. Immunology 2017, 152, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, L.; Mahajan, S.; Capietto, A.H.; Yang, Z.; Zamani, A.; Ricci, B.; Bumpass, D.B.; Meyer, M.; Su, X.; Wang-Gillam, A.; et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J. Exp. Med. 2016, 213, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Thudi, N.K.; Martin, C.K.; Murahari, S.; Shu, S.T.; Lanigan, L.G.; Werbeck, J.L.; Keller, E.T.; McCauley, L.K.; Pinzone, J.J.; Rosol, T.J. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate 2011, 71, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.L.; Zhang, H.; Baile, S.; Ljungman, M.; Kuhstoss, S.; Keller, E.T. p21CIP-1/WAF-1 induction is required to inhibit prostate cancer growth elicited by deficient expression of the Wnt inhibitor Dickkopf-1. Cancer Res. 2010, 70, 9916–9926. [Google Scholar] [CrossRef] [Green Version]
- Rachner, T.D.; Thiele, S.; Göbel, A.; Browne, A.; Fuessel, S.; Erdmann, K.; Wirth, M.P.; Fröhner, M.; Todenhöfer, T.; Muders, M.H.; et al. High serum levels of Dickkopf-1 are associated with a poor prognosis in prostate cancer patients. BMC Cancer 2014, 14, 649. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Wise, D.R.; Schneider, J.A.; Armenia, J.; Febles, V.A.; McLaughlin, B.; Brennan, R.; Thoren, K.L.; Abida, W.; Sfanos, K.S.; De Marzo, A.M.; et al. Dickkopf-1 Can Lead to Immune Evasion in Metastatic Castration-Resistant Prostate Cancer. JCO Precision. Oncol. 2020, 4, 1167–1179. [Google Scholar] [CrossRef]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [Green Version]
- Morishita, M.; di Luccio, E. Cancers and the NSD family of histone lysine methyltransferases. Biochim. Biophys. Acta 2011, 1816, 158–163. [Google Scholar] [CrossRef]
- Yang, P.; Guo, L.; Duan, Z.J.; Tepper, C.G.; Xue, L.; Chen, X.; Kung, H.J.; Gao, A.C.; Zou, J.X.; Chen, H.W. Histone methyltransferase NSD2/MMSET mediates constitutive NF-kappaB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell Biol. 2012, 32, 3121–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Want, M.Y.; Tsuji, T.; Singh, P.K.; Thorne, J.L.; Matsuzaki, J.; Karasik, E.; Gillard, B.; Cortes Gomez, E.; Koya, R.C.; Lugade, A.; et al. WHSC1/NSD2 regulates immune infiltration in prostate cancer. J. Immunother. Cancer 2021, 9, e001374. [Google Scholar] [CrossRef] [PubMed]
- Want, M.Y.; Karasik, E.; Gillard, B.; McGray, A.J.R.; Battaglia, S. Inhibition of WHSC1 Allows for Reprogramming of the Immune Compartment in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 8742. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug. Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef]
- Quatrini, L.; Della Chiesa, M.; Sivori, S.; Mingari, M.C.; Pende, D.; Moretta, L. Human NK cells, their receptors and function. Eur. J. Immunol. 2021, 51, 1566–1579. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Granjeaud, S.; Guerin, M.; Thomassin-Piana, J.; Rocchi, P.; Salem, N.; Walz, J.; Moretta, A.; Olive, D. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015, 6, 14360–14373. [Google Scholar] [CrossRef] [Green Version]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [Green Version]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef]
- Demaria, O.; Gauthier, L.; Debroas, G.; Vivier, E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur. J. Immunol. 2021, 51, 1934–1942. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. Could Harnessing Natural Killer Cell Activity Be a Promising Therapy for Prostate Cancer? Crit. Rev. Immunol. 2021, 41, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Crespo, M.; Gurel, B.; Dolling, D.; Rekowski, J.; Sharp, A.; Petremolo, A.; Sumanasuriya, S.; Rodrigues, D.N.; Ferreira, A.; et al. CD38 in Advanced Prostate Cancers. Eur. Urol. 2021, 79, 736–746. [Google Scholar] [CrossRef]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100.e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucali, P.A.; Lin, C.C.; Carthon, B.C.; Bauer, T.M.; Tucci, M.; Italiano, A.; Iacovelli, R.; Su, W.C.; Massard, C.; Saleh, M.; et al. Targeting CD38 and PD-1 with isatuximab plus cemiplimab in patients with advanced solid malignancies: Results from a phase I/II open-label, multicenter study. J. Immunother. Cancer 2022, 10, e003697. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Han, H.H.; Wang, Y.; Zhang, B.; Zhou, B.; Cheng, Y.; Rumandla, A.; Gurrapu, S.; Chakraborty, G.; Su, J.; et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell 2019, 36, 139–155.e110. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Roh, M.; Naseem, A.F.; Lysy, B.; Desouki, M.M.; Unno, K.; Abdulkadir, S.A. Bmi1 marks distinct castration-resistant luminal progenitor cells competent for prostate regeneration and tumour initiation. Nat. Commun. 2016, 7, 12943. [Google Scholar] [CrossRef] [Green Version]
- Clermont, P.L.; Lin, D.; Crea, F.; Wu, R.; Xue, H.; Wang, Y.; Thu, K.L.; Lam, W.L.; Collins, C.C.; Wang, Y.; et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenet. 2015, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, P.L.; Crea, F.; Chiang, Y.T.; Lin, D.; Zhang, A.; Wang, J.Z.; Parolia, A.; Wu, R.; Xue, H.; Wang, Y.; et al. Identification of the epigenetic reader CBX2 as a potential drug target in advanced prostate cancer. Clin. Epigenet. 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Jiao, D.; Han, D.; Wu, J.; Wei, F.; Zheng, G.; Guo, Z.; Xi, W.; Yang, F.; Xie, P.; et al. Knockdown of RNF2 induces cell cycle arrest and apoptosis in prostate cancer cells through the upregulation of TXNIP. Oncotarget 2017, 8, 5323–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayam, R.M.; Sun, C.X.; Choy, C.H.; Mancuso, G.; Glogauer, M.; Botelho, R.J. The Lipid Kinase PIKfyve Coordinates the Neutrophil Immune Response through the Activation of the Rac GTPase. J. Immunol. 2017, 199, 2096–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Xu, Y.; Cheung, A.K.; Tomlinson, R.C.; Alcazar-Roman, A.; Murphy, L.; Billich, A.; Zhang, B.; Feng, Y.; Klumpp, M.; et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 2013, 20, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Xu, Y.; Kim, Y.M.; Loureiro, J.; Huang, Q. PIKfyve, a class III lipid kinase, is required for TLR-induced type I IFN production via modulation of ATF3. J. Immunol. 2014, 192, 3383–3389. [Google Scholar] [CrossRef] [Green Version]
- Min, S.H.; Suzuki, A.; Stalker, T.J.; Zhao, L.; Wang, Y.; McKennan, C.; Riese, M.J.; Guzman, J.F.; Zhang, S.; Lian, L.; et al. Loss of PIKfyve in platelets causes a lysosomal disease leading to inflammation and thrombosis in mice. Nat. Commun. 2014, 5, 4691. [Google Scholar] [CrossRef] [Green Version]
- Hudkins, R.L.; Becknell, N.C.; Zulli, A.L.; Underiner, T.L.; Angeles, T.S.; Aimone, L.D.; Albom, M.S.; Chang, H.; Miknyoczki, S.J.; Hunter, K.; et al. Synthesis and biological profile of the pan-vascular endothelial growth factor receptor/tyrosine kinase with immunoglobulin and epidermal growth factor-like homology domains 2 (VEGF-R/TIE-2) inhibitor 11-(2-methylpropyl)-12,13-dihydro-2-methyl-8-(pyrimidin-2-ylamino)-4H-indazolo[5, 4-a]pyrrolo[3,4-c]carbazol-4-one (CEP-11981): A novel oncology therapeutic agent. J. Med. Chem. 2012, 55, 903–913. [Google Scholar] [CrossRef]
- Qiao, Y.; Choi, J.E.; Tien, J.C.; Simko, S.A.; Rajendiran, T.; Vo, J.N.; Delekta, A.D.; Wang, L.; Xiao, L.; Hodge, N.B.; et al. Autophagy Inhibition by Targeting PIKfyve Potentiates Response to Immune Checkpoint Blockade in Prostate Cancer. Nat. Cancer 2021, 2, 978–993. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Clinical Trial Number and Trial Phase | Description | Results (OS in Months; PSA in ng/mL) | Ref. | |
|---|---|---|---|---|
| Sipuleucel-T (NCT00065442) | III | Active cellular (peripheral-blood mononuclear and antigen-presenting cells) | OS, 25.8 with Spiuleucel-T 21.7 with placebo; PSA, 51.7 with Sipuleucel-T 47.2 with placebo | [27] |
| PROSTVAC (EudraCT 2010-021196-85) | III | Recombinant vaccinia and fowlpox viruses containing transgenes for human PSA and 3 T-cell costimulatory molecules | OS, 23.1 with viral vectors 22.8 with placebo; PSA, 71.4 with viral vectors 82.6 with placebo | [33] |
| VANCE (NCT02390063) | I | Replication-deficient viruses targeting oncofetal self-antigen 5T4 (early-stage PCa) | PSA, >100% increase in PSA levels post-vaccination for 3 participants, with others showing <50% increase | [34] |
| KRM-20 (UMIN000011028) | II | KRM-20 is a 20 peptide mix that induces cytotoxic T-lymphocytes against 12 tumor-associated antigens | No significant difference in PSA response, but both human leukocyte antigen (HLA)-IgG and CTL responses increased in KRM-20 arm | [35] |
| Blood-derived dendritic cells (DCs) (NCT02692976) | IIa | Monotherapies or combinations of myeloid DCs and/or plasmacytoid DCs used to induce cytotoxic T cells (intranodal injection) | Radiographic progression-free survival (rPFS) found to be 18.8 months in those with functional antigen-specific T cells (n = 5), and 5.1 months in those without (n = 16) | [36] |
| Anti-RhoC (NCT03199872) | I/II | The RhoC protein has been correlated with advanced cancer cells and metastasis, so this trial tests a vaccine to inhibit its function | 86% of patients had a significant T-cell response during vaccinations, and 90% during the follow-up (functional T effector memory cells were seen, but not Tregs) | [37] |
| PCD4989g Atezolizumab (NCT01375842) | I | Small-molecule atezolizumab (PD-L1 inhibitor) with previous treatment using Sipuleucel-T or enzalutamide | PSA, 8.6% response, OS, 14.7 months, overall limited efficacy, so combination approach may be needed | [38] |
| IMbassador250 Atezolizumab (NCT03016312) | III | Small-molecule atezolizumab (PD-L1 inhibitor) with previous treatment using abiraterone; concurrent with enzalutamide for both arms | Stopped early because patients were at risk of immune-mediated adverse events; OS, 15.2 months for atezolizumab + enzalutamide vs. 16.6 months for enzalutamide only | [39] |
| KEYNOTE-199 Pembrolizumab (NCT02787005) | II | Monoclonal antibody pembrolizumab (PD-1 inhibitor) with previous treatment using docetaxel or enzalutamide | PSA, <10% response, ORR, <5%, rPFS, 2.1, 2.1, and 3.7 months for 3 cohorts (Cohort 1: PD-L1-positive; Cohort 2: PD-L1-negative; Cohort 3: bone-predominant disease, regardless of PD-L1 expression) | [40] |
| STARVE-PC Ipilimumab/Nivolumab (NCT02601014) | II | Ipilimumab (anti-CTLA4 monoclonal antibody), nivolumab (PD1 inhibitor), some concurrent treatment with nivolumab (all with enzalutamide) | Lower alkaline phosphatase levels in a subset of patients treated with immune blockade; did not meet primary endpoint | [41] |
| CheckMate650 Ipilimumab (NCT02985957) | II | Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor); one subset treated with cabazitaxe. Concurrent with nivolumab and higher dose ipilimumab | OS, 15.2 months in post-chemo cohort and 19 months in pre-chemo; ORR, 10% in post-chemo cohort and 25% in pre-chemo | [42] |
| MDX-010 Ipilimumab (NCT00323882) | I/II | Ipilimumab (CTLA4 inhibitor), dose-escalation treatments of ipilimumab combined with radiotherapy | PSA, 8 patients had PSA decline >50% and 1 had a complete response; high dose of 10 mg/kg ipilimumab showed a manageable safety profile | [43] |
| Trial Name and Trial Phase | Treatment(s) | Purpose and Expected Completion Date | ||
|---|---|---|---|---|
| CHOMP (NCT04104893) | II | Pembrolizumab (PD-1 inhibitor) | To evaluate the activity and efficacy of pembrolizumab in mismatch repair deficiency (dMMR) and CDK12 biallelic inactivation mCPRC patients | 3/2023 |
| PERSEUS1 (NCT03506997) | II | Pembrolizumab (PD-1 inhibitor) | To evaluate the efficacy of pembrolizumab. To determine PD-1 and PD-L1, Treg infiltration, CD3, CD8, and lymphocyte infiltration | 9/2025 |
| NCT03406858 | II | Pembrolizumab (PD-1 inhibitor), HER2Bi-armed activated T cells | To test if the combination of the HER2Bi-armed T cells and pembrolizumab is better at treating mCRPC patients | 12/2021 (Active) |
| INSPIRE (NCT04717154) | II | Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor) | To evaluate the effects of 4 cycles of combination treatments (ipilimumab and nivolumab), followed by monotherapy nivolumab in participants with mCPPC | 6/2025 |
| IMPACT (NCT03570619) | II | Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor) | To evaluate the efficacy of combo treatment in patients with mCRPC and CDK12 mutations | 5/2023 |
| NCT03456804 | II | ESK981 (Pan-VEGFR/TIE2 tyrosine kinase inhibitor and PIKfyve lipid kinase inhibitor) | To study the side effects and how well ESK981 works in treating patients with mCRPC | 10/2022 |
| NCT03792841 | I | Acapatamab (bispecific T-cell engager), pembrolizumab (PD-1 inhibitor) | To determine the max tolerated dose of Acapatamab (a half-life extended (HLE) bispecific T-cell engager (BiTE®) construct) alone and in combination with pembrolizumab | 6/2025 |
| NCT05293496 | I | MGC018 (CD276 inhibitor), lorigerlimab (dual PD-1 × CTLA-4 inhibitors) | To determine the safety and efficacy of MGC018 + lorigerlimab combo treatment | 3/2025 |
| NCT05177770 | II | SRF617 (CD39 inhibitor), etrumadenant (dual A2aR/A2bR antagonist), zimberelimab (PD-1 inhibitor) | To evaluate the safety and efficacy of SRF617 in combination with etrumadenant and zimberelimab | 11/2023 |
| IceCAP (NCT03673787) | I/II | Ipatasertib (AKT inhibitor), atezolizumab (PD-L1 inhibitor) | Proof of concept for the combination of ipatasertib and atezolizumab acting on PI3K hyperactivated tumors | 11/2023 |
| NCT03061539 | II | Nivolumab (PD1 inhibitor), ipilimumab (CTLA4 inhibitor) | To evaluate the efficacy of PD-1 inhibitor in combination with CTLA4 inhibitor | 7/2025 |
| NCT02933255 | I/II | PROSTVAC-V/F (vaccine), nivolumab (PD1 inhibitor) | To evaluate the combination therapy of PROSTVAC and nivolumab for safety and effectiveness | 8/2022 |
| Rad2Nivo (NCT04109729) | Ib/II | Nivolumab (PD1 inhibitor), radium-223 (radioactive isotope) | To assess the safety of this combination treatment, then expand into a phase II cohort | 4/2025 |
| NCT04159896 | II | ESK981 (multi-tyrosine kinase inhibitors), nivolumab (PD1 inhibitor) | To evaluate the safety and efficacy of these drugs in combination (ESK981 = pan-VEGFR/TIE2 tyrosine kinase inhibitor) | 3/2022 (Active) |
| NCT03651271 | II | Nivolumab (PD1 inhibitor), ipilimumab (CTLA4 inhibitor) | To evaluate treatment outcomes for patients with low vs. high levels of CD8 cells in tumor biopsy in monotherapies of nivolumab or combo | 5/2023 |
| CheckMate 7DXNCT04100018 | III | Nivolumab (PD1 inhibitor), prednisone, docetaxel | To assess the safety and efficacy of nivolumab + docetaxel in comparison to placebo + docetaxel | 8/2027 |
| NCT05169684 | II | BMS986218 (CTLA4 inhibitor), docetaxel, nivolumab (PD1 inhibitor) | To assess the safety and efficacy of BMS986218 in different combos with nivolumab and docetaxel | 2/2026 |
| PORTER (NCT03835533) | I | NKTR-214 (CD122-preferential IL2 pathway agonist), nivolumab (PD1 inhibitor), SBRT (radiation), CDX-301 (FLT3 ligand, a dendritic cell mobilizer), INO-5151 (combination of DNA plasmids encoding IL-12 and PSA/PSMA) | To evaluate the safety and efficacy of immunotherapy combinations. To explore immune biomarker response in prostate cancer after treatment with different combinations | 3/2023 |
| STELLAR-001(NCT03845166) | I | XL092 (tyrosine kinase inhibitor that targets VEGF receptors, c-Met), atezolizumab (PD-L1 inhibitor), avelumab (PD-L1 inhibitor) | To evaluate the safety, tolerability, pharmacokinetics (PK), preliminary antitumor activity by XL092 as a monotherapy or in combination with other PD-L1 inhibitors | 11/2024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Wasielewski, L.J.; Yang, J.C.; Cai, D.; Evans, C.P.; Murphy, W.J.; Liu, C. The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines 2022, 10, 1778. https://doi.org/10.3390/biomedicines10081778
Xu P, Wasielewski LJ, Yang JC, Cai D, Evans CP, Murphy WJ, Liu C. The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines. 2022; 10(8):1778. https://doi.org/10.3390/biomedicines10081778
Chicago/Turabian StyleXu, Pengfei, Logan J. Wasielewski, Joy C. Yang, Demin Cai, Christopher P. Evans, William J. Murphy, and Chengfei Liu. 2022. "The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer" Biomedicines 10, no. 8: 1778. https://doi.org/10.3390/biomedicines10081778
APA StyleXu, P., Wasielewski, L. J., Yang, J. C., Cai, D., Evans, C. P., Murphy, W. J., & Liu, C. (2022). The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines, 10(8), 1778. https://doi.org/10.3390/biomedicines10081778