
Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target?

 , , ,
, , , 
Abstract
:1. Mitochondria and Homeostasis
2. What Is the UPRmt?
3. Mitochondrial Diseases
4. Neurodegeneration
4.1. Parkinson’s Diseases
4.2. Alzheimer’s Disease
4.3. Huntington’s Disease
4.4. Amyotrophic Lateral Sclerosis
5. Heart Diseases
6. Lifespan
7. Therapeutic Concerns
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4E-BP1 | Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 |
| ABCB10 | ATP-binding cassette sub-family B member 10 |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating Transcription Factor 4 |
| ATF5 | Activating Transcription Factor 5 |
| ATFS-1 | Activated transcription factor 1 |
| ATP | adenosine triphosphate |
| Aβ | amyloid beta protein |
| cAMP | cyclic adenosine monophosphate |
| cGMP | cyclic guanosine monophosphate |
| CHOP | C/EBP homologous protein |
| CSCs | cancer stem cells |
| Eif2α | eucaryotic initiation factor 2 alpha |
| HD | Huntington’s disease |
| HSP | heat shock protein |
| HtrA2 | HtrA Serine Peptidase 2 |
| Htt | huntingtin |
| IGF-1 | insulin growth factor 1 |
| ISR | integrated stress response |
| LC3 | microtubule-associated protein-1 light chain-3 |
| LIR | LC3-interacting region |
| MELAS | mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| mtDNA | mitochondrial DNA |
| ND1 | NADH–ubiquinone oxidoreductase chain 1 |
| NFT | neurofibrillary tangles |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| OXPHOS | oxidation phosphorylation |
| P-Eif2α | Phosphorylated Eukaryotic Initiation Factor 2 alpha |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced kinase 1 |
| PITRM1 | pitrilysin metallopeptidase 1 |
| ROS | reactive oxygen species |
| SIRT | sirtuin |
| TDP-43 | TAR DNA-binding protein 43 |
| UPRmt | mitochondrial unfolded protein response |
References
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Jo, S.-H.; Kim, M.-Y.; Kim, T.-H.; Ahn, Y.-H. Role of transcription factor acetylation in the regulation of metabolic homeostasis. Protein Cell 2015, 6, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Danziger, J.; Zeidel, M.L. Osmotic homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 852–862. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fernández, M.R.; Valpuesta, J.M. Hsp70 chaperone: A master player in protein homeostasis. F1000Research 2018, 7, 1497. [Google Scholar] [CrossRef] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Hederstedt, L. Heme A biosynthesis. Biochim. Biophys. Acta 2012, 1817, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Nakhle, J.; Rodriguez, A.-M.; Vignais, M.-L. Multifaceted roles of mitochondrial components and metabolites in metabolic diseases and cancer. Int. J. Mol. Sci. 2020, 21, 4405. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial calcium regulation of redox signaling in cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Vertika, S.; Singh, K.K.; Rajender, S. Mitochondria, spermatogenesis, and male infertility—An update. Mitochondrion 2020, 54, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating mitophagy in mitochondrial disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef] [PubMed]
- Valenci, I.; Yonai, L.; Bar-Yaacov, D.; Mishmar, D.; Ben-Zvi, A. Parkin modulates heteroplasmy of truncated mtDNA in Caenorhabditis elegans. Mitochondrion 2015, 20, 64–70. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Muñoz-Carvajal, F.; Sanhueza, M. The mitochondrial unfolded protein response: A hinge between healthy and pathological. Aging Front. Aging Neurosci. 2020, 12, 581849. [Google Scholar] [CrossRef]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, K.; Liu, Y. UPR mt coordinates immunity to maintain mitochondrial homeostasis and animal fitness. Mitochondrion 2018, 41, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.S.; Haynes, C.M. Folding the mitochondrial UPR into the integrated stress response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Cabezas, R.; El-Bachá, R.S.; González, J.; Barreto, G.E. Mitochondrial functions in astrocytes: Neuroprotective implications from oxidative damage by rotenone. Neurosci. Res. 2012, 74, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.; Gillis, R.; Fiorese, C.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [Green Version]
- Runkel, E.D.; Liu, S.; Baumeister, R.; Schulze, E. Surveillance-activated defenses block the ros-induced mitochondrial unfolded protein response. PLoS Genet. 2013, 9, e1003346. [Google Scholar] [CrossRef] [Green Version]
- Smyrnias, I. The mitochondrial unfolded protein response and its diverse roles in cellular stress. Int. J. Biochem. Cell Biol. 2021, 133, 105934. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. JBIC J. Biol. Inorg. Chem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell. 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [Green Version]
- Naresh, N.U.; Haynes, C.M. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033944. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Palam, L.R.; Jiang, L.; Narasimhan, J.; Staschke, K.A.; Wek, R.C. Phosphorylation of eIF2 Directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 2008, 283, 7064–7073. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Angelastro, J.M.; Merino, D.; Zhou, Q.; Siegelin, M.D.; Greene, L.A. Dominant-negative ATF5 rapidly depletes survivin in tumor cells. Cell Death Dis. 2019, 10, 709. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswantahan, M.; Schoonjans, K.; et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [Green Version]
- Zeviani, M.; Carelli, V. Mitochondrial disorders. Curr. Opin. Neurol. 2007, 20, 564–571. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. 2016, 73, 4577–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1 alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Chukwudi, C.U. rRNA binding sites and the molecular mechanism of action of the tetracyclines. Antimicrob. Agents Chemother. 2016, 60, 4433–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meydan, S.; Marks, J.; Klepacki, D.; Sharma, V.; Baranov, P.V.; Firth, A.E.; Margus, T.; Kefi, A.; Vázquez-Laslop, N.; Mankin, A.S. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol. Cell 2019, 74, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res. 2008, 36, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ryu, D.; Houtkooper, R.H.; Auwerx, J. Antibiotic use and abuse: A threat to mitochondria and chloroplasts with impact on research, health, and environment. BioEssays 2015, 37, 1045–1053. [Google Scholar] [CrossRef]
- Palmeira, C.; Teodoro, J.; Amorim, J.A.; Steegborn, C.; Sinclair, D.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. Mitochondria and antibiotics: For good or for evil? Biomolecules 2021, 11, 1050. [Google Scholar] [CrossRef]
- Gao, Z.; Chen, Y.; Guan, M.-X. Mitochondrial DNA mutations associated with aminoglycoside induced ototoxicity. J. Otol. 2017, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.S.; Xie, Y.; Tor, Y. Antibiotics that target protein synthesis. Wiley Interdiscip. Rev. RNA 2011, 2, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-González, A.; Gómez-Fernandez, D.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in combination with mitochondrial cofactors improve mitochondrial function in cellular models of mitochondrial diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef]
- Lange, K.W.; Li, S. Resveratrol, pterostilbene, and dementia. Biofactors 2018, 44, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Li, S.; Lin, C.-C. Effect of resveratrol and pterostilbene on aging and longevity. BioFactors 2018, 44, 69–82. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Liu, X.; Li, X.; Cao, Y.; Bai, Y.; Qi, F. Pterostilbene inhibits amyloid-beta-induced neuroinflammation in a microglia cell line by inactivating the NLRP3/caspase-1 inflammasome pathway. J. Cell Biochem. 2018, 119, 7053–7062. [Google Scholar] [CrossRef] [PubMed]
- Natalin, H.M.; Garcia, A.F.E.; Ramalho, L.N.Z.; Restini, C.B.A. Resveratrol improves vasoprotective effects of captopril on aortic remodeling and fibrosis triggered by renovascular hypertension. Cardiovasc. Pathol. 2016, 25, 116–119. [Google Scholar] [CrossRef]
- Malik, S.A.; Acharya, J.D.; Mehendale, N.K.; Kamat, S.S.; Ghaskadbi, S.S. Pterostilbene reverses palmitic acid mediated insulin resistance in HepG2 cells by reducing oxidative stress and triglyceride accumulation. Free Radic. Res. 2019, 53, 815–827. [Google Scholar] [CrossRef]
- Zhou, J.; Ci, X.; Ma, X.; Yu, Q.; Cui, Y.; Zhen, Y.; Li, S. Pterostilbene Activates the Nrf2-Dependent Antioxidant Response to Ameliorate Arsenic-Induced Intracellular Damage and Apoptosis in Human Keratinocytes. Front. Pharmacol. 2019, 10, 497. [Google Scholar] [CrossRef]
- Beach, T.G. A review of biomarkers for neurodegenerative disease: Will they swing us across the valley? Neurol. Ther. 2017, 6 (Suppl. 1), 5–13. [Google Scholar] [CrossRef] [Green Version]
- Evidente, V.G.H.; Adler, C.H.; Sabbagh, M.N.; Connor, D.J.; Hentz, J.G.; Caviness, J.N.; Sue, L.I.; Beach, T.G. Neuropathological findings of PSP in the elderly without clinical PSP: Possible incidental PSP? Parkinsonism Relat. Disord. 2011, 17, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigerio, R.; Fujishiro, H.; Ahn, T.B.; Josephs, K.A.; Maraganore, D.M.; DelleDonne, A.; Parisi, J.E.; Klos, K.J.; Boeve, B.F.; Disckson, D.W.; et al. Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol. Aging 2011, 32, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsnelson, A.; De Strooper, B.; Zoghbi, H.Y. Neurodegeneration: From cellular concepts to clinical applications. Sci. Transl. Med. 2016, 8, 364. [Google Scholar] [CrossRef]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.; Abramov, A.; Plun-Favreau, H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part I. Expert Opin. Ther. Targets 2010, 14, 369–385. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and Disease Specificity of NADH CoQ1 Reductase (Complex I) Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Tohgi, H.; Yonezawa, H.; Takahashi, S.; Sato, N.; Kato, E.; Kudo, M.; Hatano, K.; Sasaki, T. Cerebral blood flow and oxygen metabolism in senile dementia of Alzheimer’s type and vascular dementia with deep white matter changes. Neuroradiology 1998, 40, 131. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic. Res. 2012, 46, 1313–1326. [Google Scholar] [CrossRef]
- Parker, W.D.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yu, S.; Wang, J.; Qiao, J.; Liu, Y.; Wang, S.; Zhao, Y. Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in Drosophila melanogaster PINK1 model of Parkinson’s disease. J. Ethnopharmacol. 2020, 247, 112213. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Pham, M.T.; Mitchell, M.B.; Yeom, S.G.; Jordan, S.; Chang, A.; Sopher, B.L.; La Spada, A.R. 4E-BP1 Protects neurons from misfolded protein stress and Parkinson’s disease toxicity by inducing the mitochondrial unfolded protein response. J. Neurosci. 2020, 40, 8734–8745. [Google Scholar] [CrossRef]
- Imai, Y.; Gehrke, S.; Wang, H.-Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–3111. [Google Scholar] [CrossRef] [Green Version]
- 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef]
- Grossberg, G.T.; Tong, G.; Burke, A.D.; Tariot, P.N. Present algorithms and future treatments for Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 67, 1157–1171. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal 2012, 16, 1434–1455. [Google Scholar] [CrossRef]
- Kish, S.J.; Mastrogiacomo, F.; Guttman, M.; Furukawa, Y.; Taanman, J.W.; Dozic, S.; Pandolfo, M.; Lamarche, J.; DiStefano, L.; Chang, L.-J. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: A nonspecific change? J. Neurochem. 1999, 72, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.O.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, W.A.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psych. 2021, 26, 5733–5750. [Google Scholar] [CrossRef]
- Shen, Y.; Ding, M.; Xie, Z.; Liu, X.; Yang, H.; Jin, S.; Xu, S.; Zhu, Z.; Wang, Y.; Wang, D.; et al. Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell. Neurosci. 2019, 13, 568. [Google Scholar] [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Landles, C.; Bates, G.P. Huntingtin and the molecular pathogenesis of Huntington’s disease: Fourth in molecular medicine review series. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Fukui, H.; Wang, X.; Pinto, M.; Moraes, C.T. The Striatum Is Highly Susceptible to Mitochondrial Oxidative Phosphorylation Dysfunctions. J. Neurosci. 2011, 31, 9895–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, M.; Qi, X.; Guo, X. Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim. Biophys. Acta—Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Yano, M. ABCB10 depletion reduces unfolded protein response in mitochondria. Biochem. Biophys. Res. Commun. 2017, 486, 465–469. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Jawaid, A.; Khan, R.; Polymenidou, M.; Schulz, P.E. Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol. Neurodegener. 2018, 13, 63. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of ALS SOD1(G93A) mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Germain, D. Cross talk between SOD1 and the mitochondrial UPR in cancer and neurodegeneration. Mol. Cell. Neurosci. 2019, 98, 12–18. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Nandi, S.S.; Katsurada, K.; Sharma, N.M.; Mahata, S.K.; Patel, K.P. Abstract 15288: Mitochondrial Injury in Cardiomyopathy of Neurogenic Hypertension: Role of MiR-18a-5p/HIF-1a Axis. Circulation 2020, 142 (Suppl. 3), A15288. [Google Scholar] [CrossRef]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 2015, 524, 485–488. [Google Scholar] [CrossRef]
- García-Díaz, L.; Coserria, F.; Antiñolo, G. Hypertrophic Cardiomyopathy due to Mitochondrial Disease: Prenatal Diagnosis, Management, and Outcome. Case Rep. Obstet. Gynecol. 2013, 2013, 472356. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; Gonzalez, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jasper, H.; Toan, S.; Muid, D.; Chang, X.; Zhou, H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021, 45, 102049. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.W.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, K.; Fukumoto, K.; Murakami, T.; Harada, S.I.; Hosono, R.; Wadhwa, R.; Mitsui, Y.; Ohkuma, S. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002, 516, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Flurkey, K.; Papaconstantinou, J.; Miller, R.A.; Harrison, D.E. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl. Acad. Sci. USA 2001, 98, 6736–6741. [Google Scholar] [CrossRef] [Green Version]
- Ozkurede, U.; Miller, R.A. Improved mitochondrial stress response in long-lived Snell dwarf mice. Aging Cell 2019, 18, e13030. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zhang, Y.J.; Cai, Y.; Xu, M.H. The role of mitochondria in mTOR-regulated longevity. Biol. Rev. Camb. Philos. Soc. 2015, 90, 167–181. [Google Scholar] [CrossRef]
- Jensen, M.B.; Jasper, H. Mitochondrial Proteostasis in the Control of Aging and Longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The Matrix Peptide Exporter HAF-1 Signals a Mitochondrial UPR by Activating the Transcription Factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.-L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrends, C.; Langer, C.A.; Boteva, R.; Böttcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin TRiC Promotes the Assembly of polyQ Expansion Proteins into Nontoxic Oligomers. Mol. Cell 2006, 23, 887–897. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neuro-degeneration in C. elegans models of Parkinson’s disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.-W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.-Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 1, 4639. [Google Scholar] [CrossRef]
- Angeli, S.; Foulger, A.; Chamoli, M.; Peiris, T.H.; Gerencser, A.; Shahmirzadi, A.A.; Andersen, J.; Lithgow, G. The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. eLife 2021, 10, 63453. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial stress response and cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Conway de Macario, E.; Marasà, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.L.; De Macario, E.C. Chaperonopathies by defect, excess, or mistake. Ann. N. Y. Acad. Sci. 2007, 1113, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chen, P.M.; Liu, C.J.; Teng, S.-C.; Wu, K.J. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis 2009, 30, 1049–1057. [Google Scholar] [CrossRef]
- Piselli, P.; Vendetti, S.; Vismara, D.; Cicconi, R.; Poccia, F.; Colizzi, V.; Delpino, A. Different expression of CD44, ICAM-1, and HSP60 on primary tumor and metastases of a human pancreatic carcinoma growing in scid mice. Anticancer Res 2000, 20, 825–831. [Google Scholar]
- O’Barazi, H.; Zhou, L.; Templeton, N.S.; Krutzsch, H.C.; Roberts, D.D. Identification of heat shock protein 60 as a molecular mediator of alpha 3 beta 1 integrin activation. Cancer Res. 2002, 62, 1541–1548. [Google Scholar]
- Wang, G.; Fan, Y.; Cao, P.; Tan, K. Insight into the mitochondrial unfolded protein response and cancer: Opportunities and challenges. Cell Biosci. 2022, 12, 1–23. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Du, F.-Y.; Zhou, Q.-F.; Sun, W.-J.; Chen, G.-L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 2019, 11, 398–420. [Google Scholar] [CrossRef]
- Gu, L.-F.; Chen, J.-Q.; Lin, Q.-Y.; Yang, Y.-Z. Roles of mitochondrial unfolded protein response in mammalian stem cells. World J. Stem Cells 2021, 13, 737–752. [Google Scholar] [CrossRef]

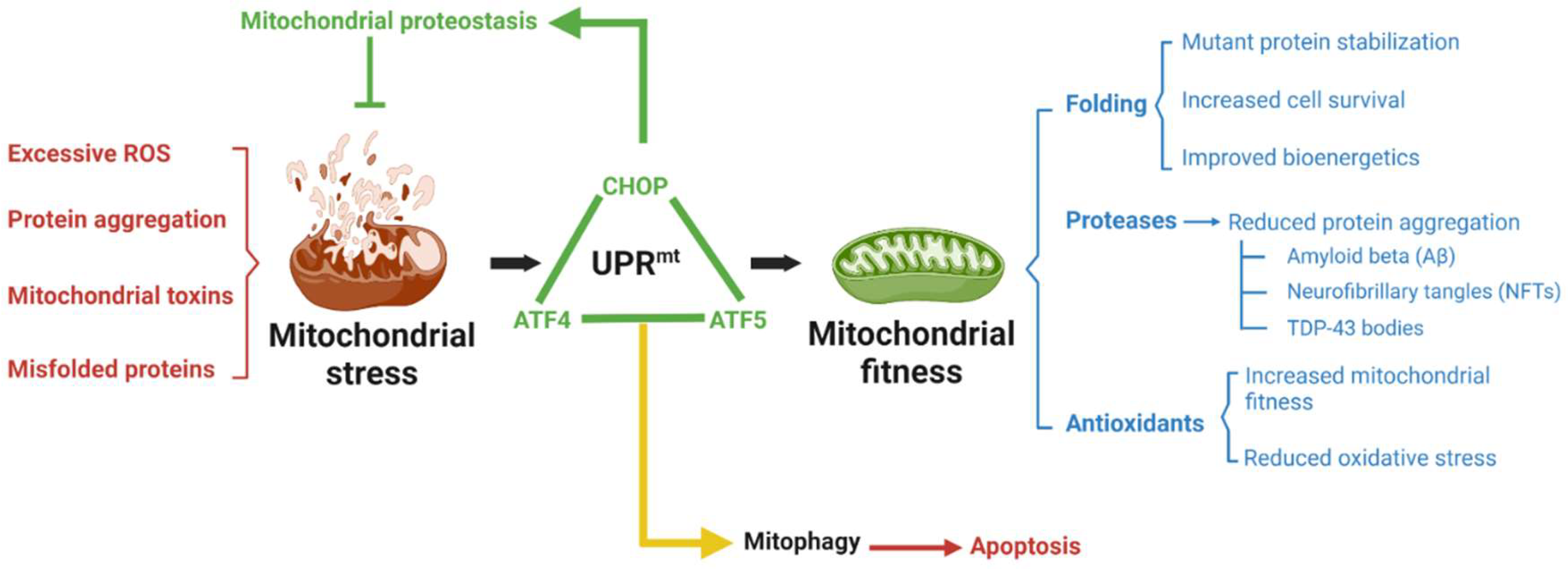

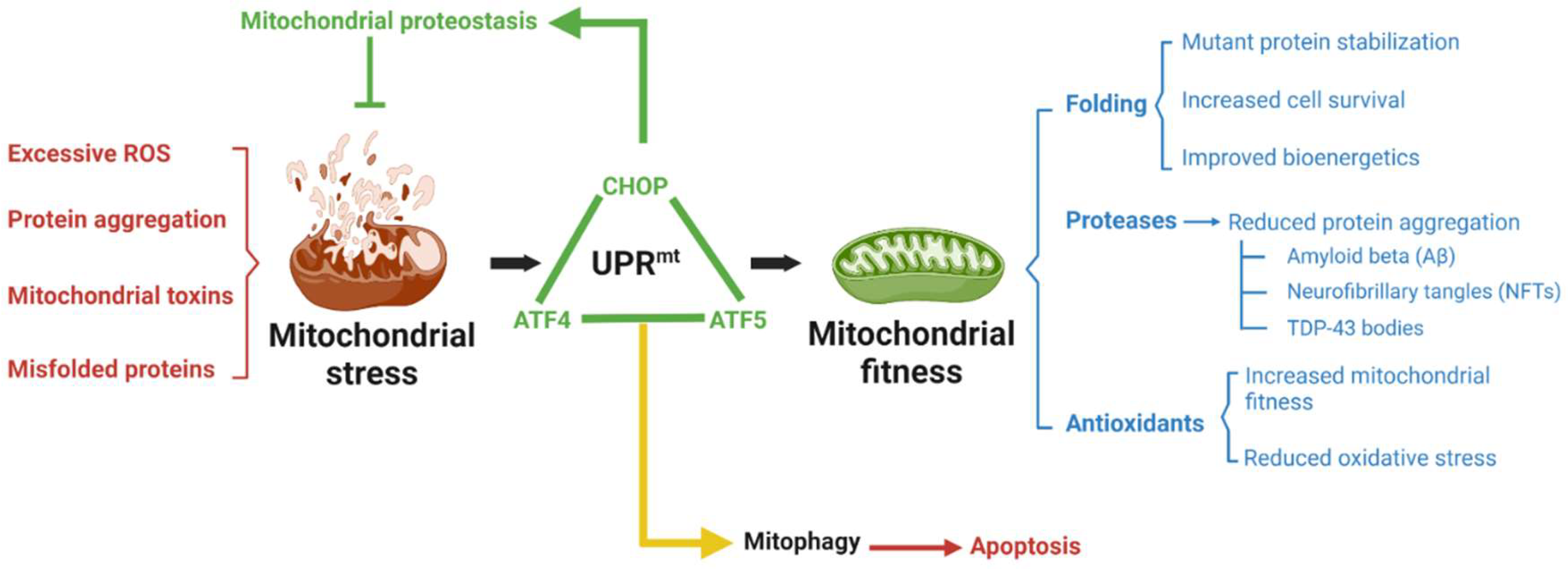
{kind=link}
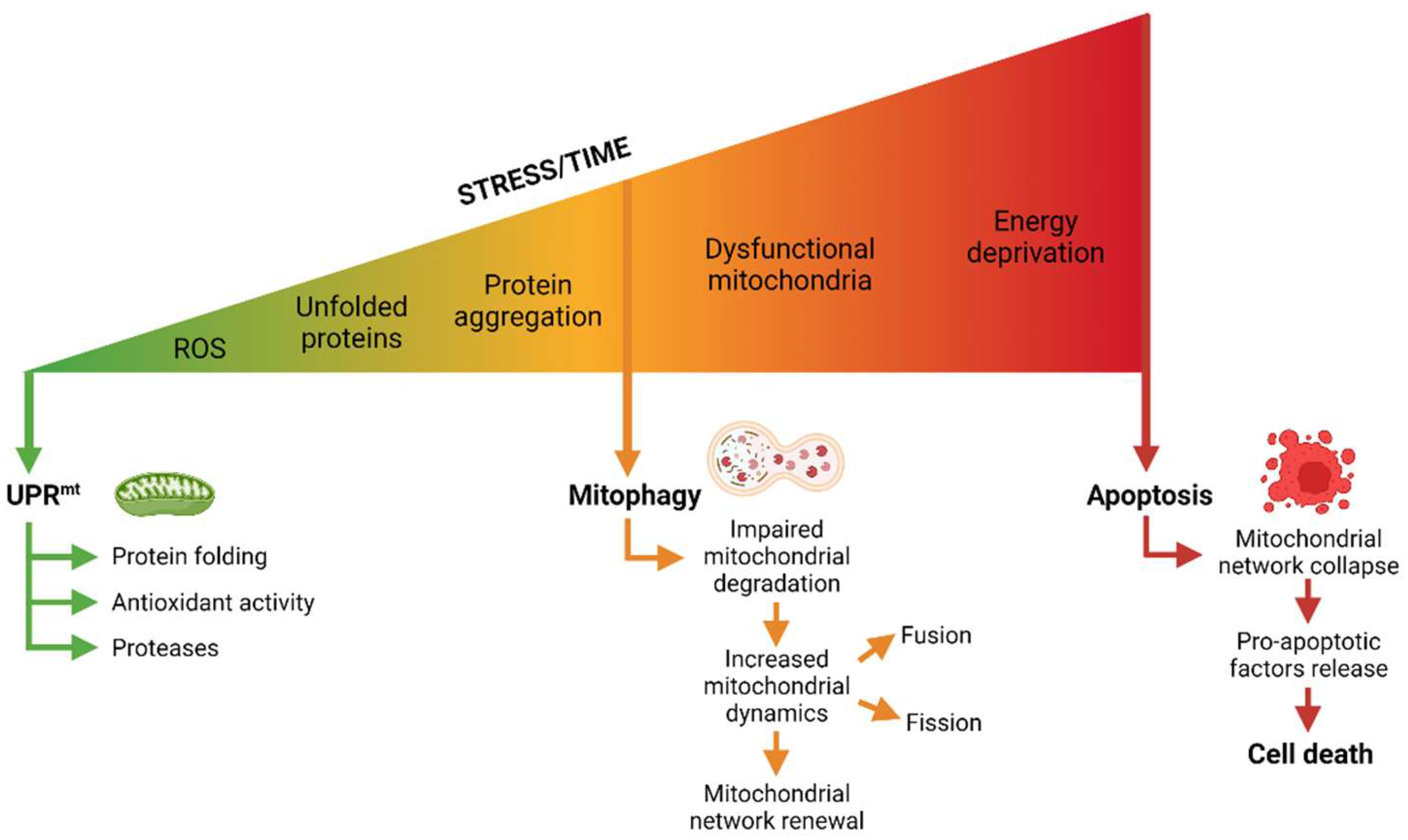
{kind=link}
| Condition | Related Studies |
|---|---|
| Mitochondrial diseases | [60,64,73] |
| Parkinson’s disease | [93,94,95,96,148] |
| Alzheimer’s disease | [105,106,107,108,109] |
| Huntington’s disease | [115] |
| Amyotrophic lateral sclerosis | [121,122,123,124] |
| Heart diseases | [132] |
| Aging | [139,140,141,142] |
| Cancer | [157,160] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10, 1611. https://doi.org/10.3390/biomedicines10071611
Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M, Reche-López D, Cilleros-Holgado P, et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines. 2022; 10(7):1611. https://doi.org/10.3390/biomedicines10071611
Chicago/Turabian StyleSuárez-Rivero, Juan M., Carmen J. Pastor-Maldonado, Suleva Povea-Cabello, Mónica Álvarez-Córdoba, Irene Villalón-García, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, Manuel Munuera-Cabeza, Diana Reche-López, Paula Cilleros-Holgado, and et al. 2022. "Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target?" Biomedicines 10, no. 7: 1611. https://doi.org/10.3390/biomedicines10071611
APA StyleSuárez-Rivero, J. M., Pastor-Maldonado, C. J., Povea-Cabello, S., Álvarez-Córdoba, M., Villalón-García, I., Talaverón-Rey, M., Suárez-Carrillo, A., Munuera-Cabeza, M., Reche-López, D., Cilleros-Holgado, P., Piñero-Pérez, R., & Sánchez-Alcázar, J. A. (2022). Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines, 10(7), 1611. https://doi.org/10.3390/biomedicines10071611