
Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders

 ,
, 
 ,
,
Abstract
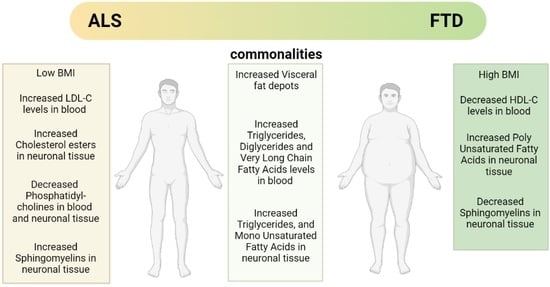
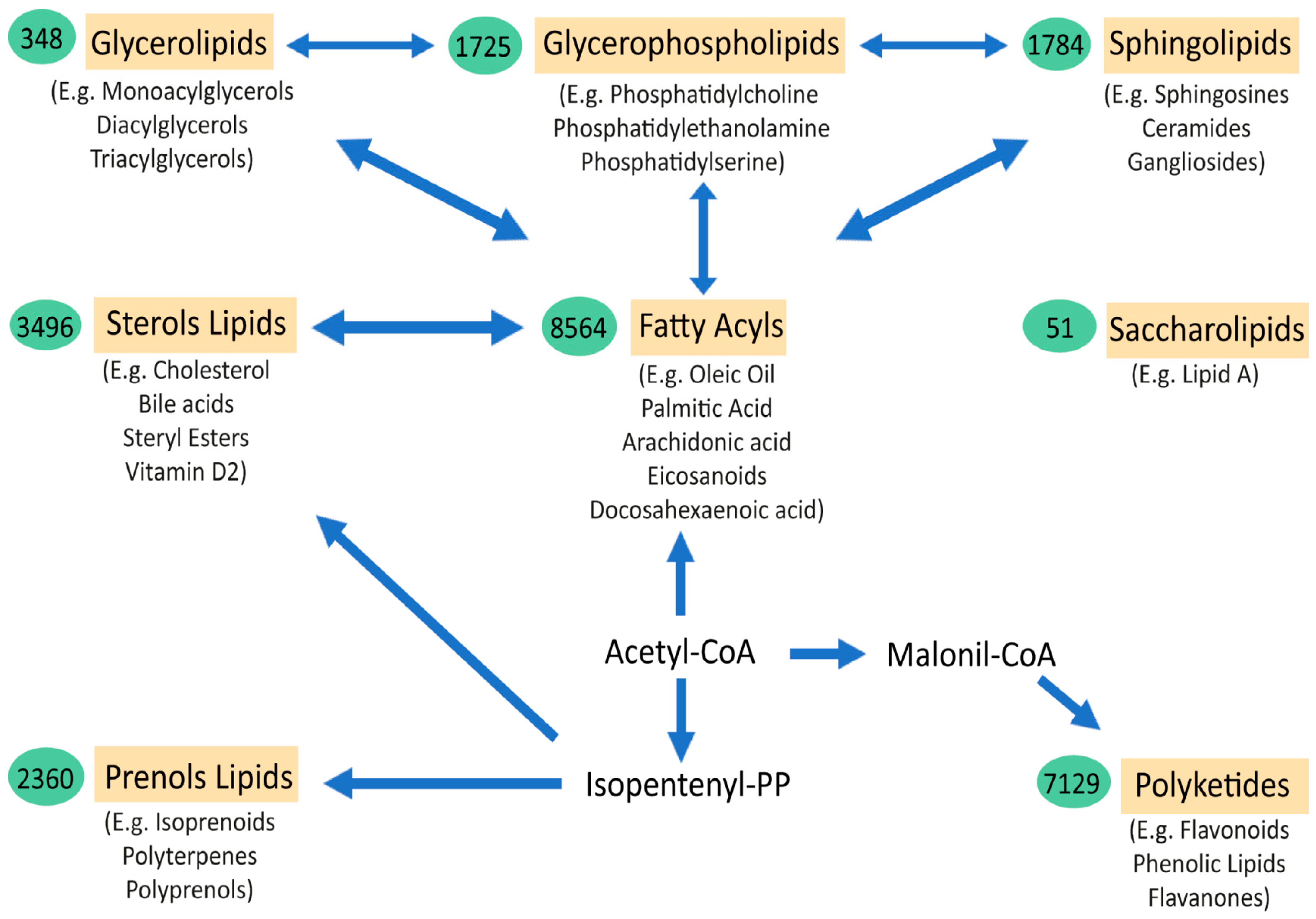
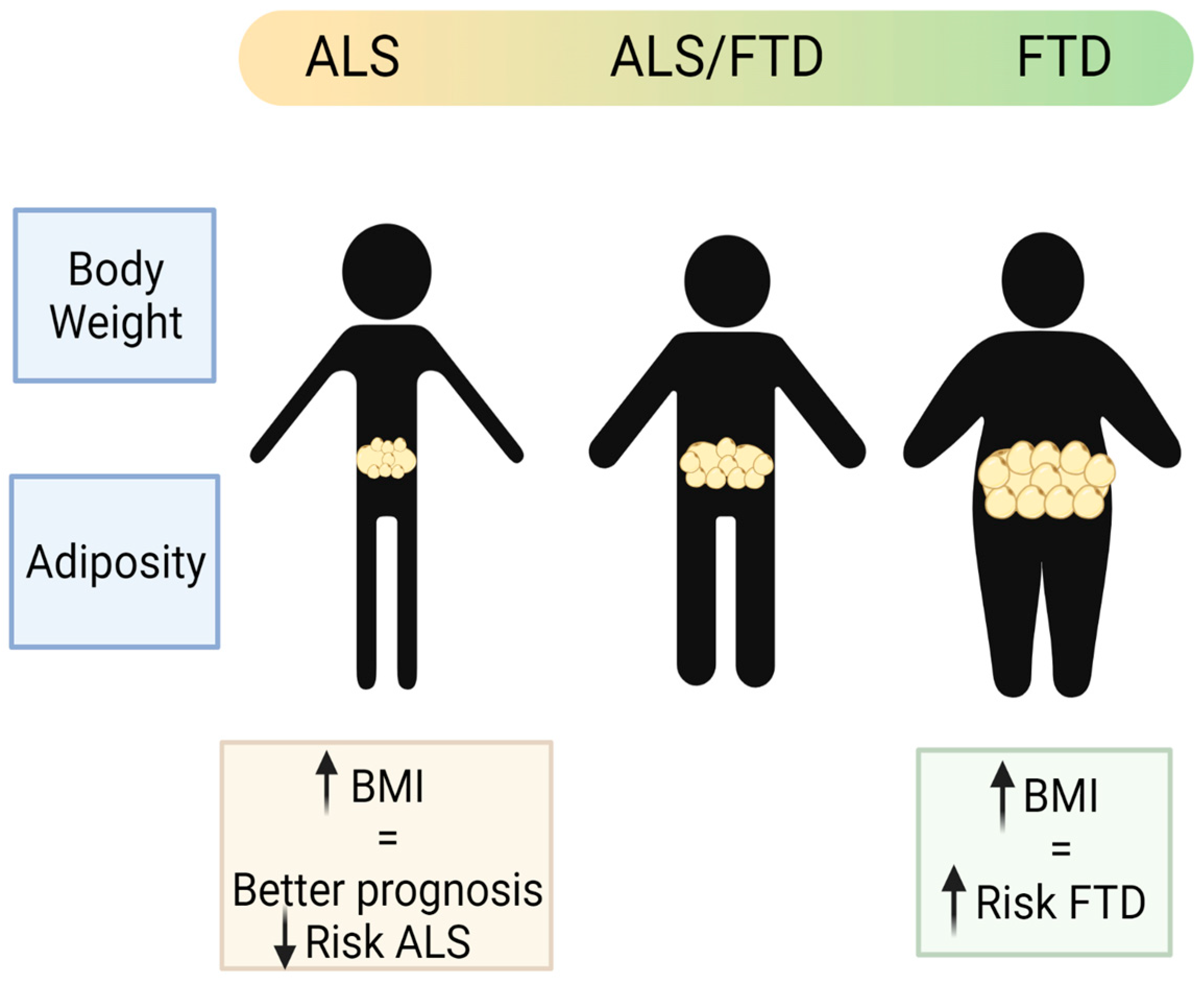
:
1. Introduction
2. Amyotrophic Lateral Sclerosis and Lipid Metabolism
2.1. Weight
2.2. Adiposity
2.3. Lipid Analysis (Classical, Lipidomic and Other -Omics Analysis)
2.3.1. Sterol Lipids
2.3.2. Glycerolipids, Fatty Acyls, and Eicosanoids
2.3.3. Sphingolipids and Ceramides
2.3.4. Glycerophospholipids
2.4. Genetics
3. FTD, Mixed Forms (ALS/FTD) and Lipid Metabolism
3.1. Weight
3.2. Adiposity
3.3. Lipid Analysis (Classical, Lipidomic, and Other -Omics Analysis)
3.4. Genetics
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 637–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghirlanda, G.; Oradei, A.; Manto, A.; Lippa, S.; Uccioli, L.; Caputo, S.; Greco, A.V.; Littarru, G.P. Evidence of Plasma CoQ10-Lowering Effect by HMG-CoA Reductase Inhibitors: A Double-Blind, Placebo-Controlled Study. J. Clin. Pharmacol. 1993, 33, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.; Mougan, I. Fatty acid composition of human brain phospholipids during normal development. J. Neurochem. 2002, 71, 2528–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-Y.; Huang, B.X.; Spector, A.A. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 2014, 56, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Saher, G.; Quintes, S.; Nave, K.-A. Cholesterol: A Novel Regulatory Role in Myelin Formation. Neuroscientist 2011, 17, 79–93. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Deguise, M.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Darios, F.; Mochel, F.; Stevanin, G. Lipids in the Physiopathology of Hereditary Spastic Paraplegias. Front. Neurosci. 2020, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- González-Guevara, E.; Cárdenas, G.; Pérez-Severiano, F.; Martínez-Lazcano, J.C. Dysregulated Brain Cholesterol Metabolism Is Linked to Neuroinflammation in Huntington’s Disease. Mov. Disord. 2020, 35, 1113–1127. [Google Scholar] [CrossRef]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Park. Dis. 2020, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchsinger, J.A.; Cheng, D.; Tang, M.X.; Schupf, N.; Mayeux, R. Central Obesity in the Elderly is Related to Late-onset Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Tolppanen, A.-M.; Ngandu, T.; Kåreholt, I.; Laatikainen, T.; Rusanen, M.; Soininen, H.; Kivipelto, M. Midlife and Late-Life Body Mass Index and Late-Life Dementia: Results from a Prospective Population-Based Cohort. J. Alzheimer’s Dis. 2013, 38, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Di Giovanni, A.; Marino, M.; Pallottini, V. Analysis of the protein network of cholesterol homeostasis in different brain regions: An age and sex dependent perspective. J. Cell. Physiol. 2012, 228, 1561–1567. [Google Scholar] [CrossRef]
- Fitzner, D.; Bader, J.M.; Penkert, H.; Bergner, C.G.; Su, M.; Weil, M.-T.; Surma, M.A.; Mann, M.; Klose, C.; Simons, M. Cell-Type- and Brain-Region-Resolved Mouse Brain Lipidome. Cell Rep. 2020, 32, 108132. [Google Scholar] [CrossRef]
- Miranda, A.M.; Bravo, F.V.; Chan, R.B.; Sousa, N.; Di Paolo, G.; Gil Oliveira, T. Differential lipid composition and regulation along the hippocampal longitudinal axis. Transl. Psychiatry 2019, 9, 144. [Google Scholar] [CrossRef]
- Jové, M.; Mota-Martorell, N.; Torres, P.; Portero-Otin, M.; Ferrer, I.; Pamplona, R. New insights into human prefrontal cortex aging with a lipidomics approach. Expert Rev. Proteom. 2021, 18, 333–344. [Google Scholar] [CrossRef]
- Yu, Q.; He, Z.; Zubkov, D.; Huang, S.; Kurochkin, I.; Yang, X.; Halene, T.; Willmitzer, L.; Giavalisco, P.; Akbarian, S.; et al. Lipidome alterations in human prefrontal cortex during development, aging, and cognitive disorders. Mol. Psychiatry 2018, 25, 2952–2969. [Google Scholar] [CrossRef] [Green Version]
- Speakman, J.R.; Westerterp, K.R. Associations between energy demands, physical activity, and body composition in adult humans between 18 and 96 y of age. Am. J. Clin. Nutr. 2010, 92, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P.; et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2020, 17, 104–118. [Google Scholar] [CrossRef]
- Lamas, N.J.; Roybon, L. Harnessing the Potential of Human Pluripotent Stem Cell-Derived Motor Neurons for Drug Discovery in Amyotrophic Lateral Sclerosis: From the Clinic to the Laboratory and Back to the Patient. Front. Drug Discov. 2021, 1. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiò, A.; Mazzini, L.; Mora, G. Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2020, 167, 107986. [Google Scholar] [CrossRef] [PubMed]
- Faber, R.; Neary, D. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1999, 53, 1158. [Google Scholar] [CrossRef]
- Seltman, R.E.; Matthews, B.R. Frontotemporal Lobar Degeneration. CNS Drugs 2012, 26, 841–870. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Saánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Pytel, V.; Hernández-Lorenzo, L.; Torre-Fuentes, L.; Sanz, R.; González, N.; Cabrera-Martín, M.N.; Delgado-Álvarez, A.; Gómez-Pinedo, U.; Matías-Guiu, J.; Matias-Guiu, J.A. Whole-Exome Sequencing and C9orf72 Analysis in Primary Progressive Aphasia. J. Alzheimer’s Dis. 2021, 80, 985–990. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Huynh, W.; Ahmed, R.; Mahoney, C.J.; Nguyen, C.; Tu, S.; Caga, J.; Loh, P.; Lin, C.S.-Y.; Kiernan, M.C. The impact of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2020, 20, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Tracey, T.; Kirk, S.; Steyn, F.; Ngo, S. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 2020, 112, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakayama, Y.; Matsuda, C.; Haraguchi, M.; Bokuda, K.; Ishikawa-Takata, K.; Kawata, A.; Isozaki, E. Prognostic significance of body weight variation after diagnosis in ALS: A single-centre prospective cohort study. J. Neurol. 2019, 266, 1412–1420. [Google Scholar] [CrossRef]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.-M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Van Mantgem, M.R.J.; van Eijk, R.P.; van der Burgh, H.K.; Tan, H.H.; Westeneng, H.J.; van Es, M.A.; Veldink, J.H.; van den Berg, L.H. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2020, 91, 867–875. [Google Scholar] [CrossRef]
- Moglia, C.; Calvo, A.; Grassano, M.; Canosa, A.; Manera, U.; D’Ovidio, F.; Bombaci, A.; Bersano, E.; Mazzini, L.; Mora, G.; et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J. Neurol. Neurosurg. Psychiatry 2019, 90, 666–673. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; Van Eijk, R.P.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Jésus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.-M.; Desport, J.-C.; Marin, B.; Couratier, P. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2017, 25, 97–104. [Google Scholar] [CrossRef]
- Gorges, M.; Vercruysse, P.; Müller, H.-P.; Huppertz, H.-J.; Rosenbohm, A.; Nagel, G.; Weydt, P.; Petersén, Å.; Ludolph, A.C.; Kassubek, J.; et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1033–1041. [Google Scholar] [CrossRef]
- Bayer, D.; Antonucci, S.; Müller, H.P.; Saad, R.; Dupuis, L.; Rasche, V.; Böckers, T.M.; Ludolph, A.C.; Kassubek, J.; Roselli, F. Disruption of orbitofrontal-hypothalamic projections in a murine ALS model and in human patients. Transl. Neurodegener. 2021, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- López-Gómez, J.J.; Ballesteros-Pomar, M.D.; Torres-Torres, B.; De la Maza, B.P.; Penacho-Lázaro, M.; Palacio-Mures, J.M.; Abreu-Padín, C.; López-Guzmán, A.; De Luis-Román, D.A. Malnutrition at diagnosis in amyotrophic lateral sclerosis (als) and its influence on survival: Using glim criteria. Clin. Nutr. 2020, 40, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Park, J.; Kim, Y.; Baek, H.; Kim, S.H. Association between nutritional status and disease severity using the amyotrophic lateral sclerosis (ALS) functional rating scale in ALS patients. Nutrition 2015, 31, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Wark, P.A.; Jenab, M.; Pearce, N.; Brayne, C.; Vermeulen, R.; Andersen, P.M.; Hallmans, G.; Kyrozis, A.; Vanacore, N.; et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: The EPIC cohort. Neurology 2013, 80, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, J.; Wang, M.; Adami, H.-O.; Alonso, A.; Bernstein, L.; Brandt, P.V.D.; Buring, J.; Daugherty, S.; Deapen, D.; Freedman, D.M.; et al. Prediagnostic body size and risk of amyotrophic lateral sclerosis death in 10 studies. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 396–406. [Google Scholar] [CrossRef]
- Mariosa, D.; Beard, J.; Umbach, D.M.; Bellocco, R.; Keller, J.; Peters, T.L.; Allen, K.D.; Ye, W.; Sandler, D.P.; Schmidt, S.; et al. Body Mass Index and Amyotrophic Lateral Sclerosis: A Study of US Military Veterans. Am. J. Epidemiol. 2017, 185, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Nakken, O.; Meyer, H.E.; Stigum, H.; Holmøy, T. High BMI is associated with low ALS risk. Neurology 2019, 93, e424–e432. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; René, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Kazamel, M.; McPherson, T.; McAdam, J.; Bamman, M.; Amara, A.; Smith, D.L., Jr.; King, P.H. Fat mass loss correlates with faster disease progression in amyotrophic lateral sclerosis patients: Exploring the utility of dual-energy x-ray absorptiometry in a prospective study. PLoS ONE 2021, 16, e0251087. [Google Scholar] [CrossRef]
- Ioannides, Z.A.; Steyn, F.; Henderson, R.D.; McCombe, P.A.; Ngo, S. Anthropometric measures are not accurate predictors of fat mass in ALS. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 486–491. [Google Scholar] [CrossRef]
- Lindauer, E.; Dupuis, L.; Müller, H.-P.; Neumann, H.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution Predicts Survival in Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e67783. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.-L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ingre, C.; Chen, L.; Zhan, Y.; Termorshuizen, J.; Yin, L.; Fang, F. Lipids, apolipoproteins, and prognosis of amyotrophic lateral sclerosis. Neurology 2020, 94, e1835–e1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chełstowska, B.; Barańczyk-Kuźma, A.; Kuźma-Kozakiewicz, M. Dyslipidemia in patients with amyotrophic lateral sclerosis—A case control retrospective study. Amyotroph. Lateral Scler. Front. Degener. 2020, 22, 195–205. [Google Scholar] [CrossRef]
- Huang, R.; Guo, X.; Chen, X.; Zheng, Z.; Wei, Q.; Cao, B.; Zeng, Y.; Shang, H. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 359–365. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Larrea, D.; Yun, T.; Xu, Y.; Hupf, J.; Zandkarimi, F.; Chan, R.B.; Mitsumoto, H. Lipidomics study of plasma from patients suggest that ALS and PLS are part of a continuum of motor neuron disorders. Sci. Rep. 2021, 11, 13562. [Google Scholar] [CrossRef]
- Nakamura, R.; Kurihara, M.; Ogawa, N.; Kitamura, A.; Yamakawa, I.; Bamba, S.; Sanada, M.; Sasaki, M.; Urushitani, M. Investigation of the prognostic predictive value of serum lipid profiles in amyotrophic lateral sclerosis: Roles of sex and hypermetabolism. Sci. Rep. 2022, 12, 1826–1836. [Google Scholar] [CrossRef]
- Bjornevik, K.; O’Reilly, J.; Cortese, M.; Furtado, J.D.; Kolonel, L.N.; Le Marchand, L.; Mccullough, M.L.; Paganoni, S.; Schwarzschild, M.A.; Shadyab, A.H.; et al. Pre-diagnostic plasma lipid levels and the risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 22, 133–143. [Google Scholar] [CrossRef]
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R.; et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res. 2017, 58, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef]
- Dodge, J.C.; Jensen, E.H.; Yu, J. Neutral lipid cacostasis contributes to disease pathogenesis in amyotrophic lateral sclerosis. J. Neurosci. 2020, 40, 9137–9147. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Bosch, L.V.D. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef]
- Filho, A.D.B.C.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. Int. J. Mol. Sci. 2021, 22, 79553. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Noh, M.-Y.; Kim, H.; Cheon, S.-Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.-W.; Sung, J.-J.; et al. 25-Hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodge, J.C.; Yu, J.; Sardi, S.P.; Shihabuddin, L.S. Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Ačimovič, J.; Lövgren-Sandblom, A.; Parini, P.; Andersen, P.M.; Björkhem, I. Cholesterol, Oxysterol, Triglyceride, and Coenzyme Q Homeostasis in ALS. Evidence against the Hypothesis That Elevated 27-Hydroxycholesterol Is a Pathogenic Factor. PLoS ONE 2014, 9, e113619. [Google Scholar] [CrossRef] [PubMed]
- Diekstra, F.P.; Saris, C.G.J.; Van Rheenen, W.; Franke, L.; Jansen, R.C.; Van Es, M.A.; Van Vught, P.W.J.; Blauw, H.M.; Groen, E.J.N.; Horvath, S.; et al. Mapping of Gene Expression Reveals CYP27A1 as a Susceptibility Gene for Sporadic ALS. PLoS ONE 2012, 7, e35333. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.C.; Kwak, S.G.; Park, J.-S.; Park, D. Relationship between statins and the risk of amyotrophic lateral sclerosis. Medicine 2021, 100, e26751. [Google Scholar] [CrossRef]
- Zheng, Z.; Sheng, L.; Shang, H. Statins and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2012, 14, 241–245. [Google Scholar] [CrossRef]
- Su, X.W.; Nandar, W.; Bs, E.B.N.; Simmons, Z.; Connor, J.R. Statins accelerate disease progression and shorten survival in SOD1G93A mice. Muscle Nerve 2016, 54, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Kuwajima, T.; Oakley, D.; Nikulina, E.; Hou, J.; Yang, W.S.; Lowry, E.R.; Lamas, N.J.; Amoroso, M.W.; Croft, G.; et al. Protein Prenylation Constitutes an Endogenous Brake on Axonal Growth. Cell Rep. 2016, 16, 545–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.; Dalvi, H.; Jain, H.; Rangaraj, N.; Singh, S.B.; Srivastava, S. Perspective insights of repurposing the pleiotropic efficacy of statins in neurodegenerative disorders: An expository appraisal. Curr. Res. Pharmacol. Drug Discov. 2020, 2, 100012. [Google Scholar] [CrossRef] [PubMed]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 2010, 258, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.-F.; Bruneteau, G.; Gordon, P.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.-M.; et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef]
- Ikeda, K.; Hirayama, T.; Takazawa, T.; Kawabe, K.; Iwasaki, Y. Relationships between Disease Progression and Serum Levels of Lipid, Urate, Creatinine and Ferritin in Japanese Patients with Amyotrophic Lateral Sclerosis: A Cross-Sectional Study. Intern. Med. 2012, 51, 1501–1508. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Eulate, G.; Ruiz-Sanz, J.I.; Riancho, J.; Zufiría, M.; Gereñu, G.; Fernández-Torrón, R.; Poza-Aldea, J.J.; Ondaro, J.; Espinal, J.B.; González-Chinchón, G.; et al. A comprehensive serum lipidome profiling of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 252–262. [Google Scholar] [CrossRef]
- Goutman, S.A.; Guo, K.; Savelieff, M.G.; Patterson, A.; Sakowski, S.A.; Habra, H.; Karnovsky, A.; Hur, J.; Feldman, E.L. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain 2022, awac025. [Google Scholar] [CrossRef]
- Sol, J.; Jové, M.; Povedano, M.; Sproviero, W.; Domínguez, R.; Piñol-Ripoll, G.; Romero-Guevara, R.; Hye, A.; Al-Chalabi, A.; Torres, P.; et al. Lipidomic traits of plasma and cerebrospinal fluid in amyotrophic lateral sclerosis correlate with disease progression. Brain Commun. 2021, 3, fcab143. [Google Scholar] [CrossRef]
- Hanrieder, J.; Ewing, A.G. Spatial Elucidation of Spinal Cord Lipid- and Metabolite- Regulations in Amyotrophic Lateral Sclerosis. Sci. Rep. 2014, 4, 5266. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jové, M.; Dalfó, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.J.; Park, N.Y.; Dubey, S.K.; Kim, T.; Ruan, K.; Bin Lim, S.; Park, S.-H.; Ha, S.; Kovlyagina, I.; et al. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci. 2021, 24, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Kakita, A.; Takahashi, H.; Ihara, Y.; Nobukuni, K.; Fujimura, H.; Sakoda, S.; Kobayashi, M. Increased expression and activation of cytosolic phospholipase A2 in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2010, 119, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Solomonov, Y.; Hadad, N.; Levy, R. Reduction of cytosolic phospholipase A2α upregulation delays the onset of symptoms in SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.-L.; Sidman, R.L.; et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Huebecker, M.; Blasco, H.; Keime, C.; Andres, C.; Corcia, P.; Priestman, D.A.; Platt, F.; Spedding, M.; Loeffler, J.-P. Inhibition of β-Glucocerebrosidase Activity Preserves Motor Unit Integrity in a Mouse Model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2017, 7, 5235. [Google Scholar] [CrossRef]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.-P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’H, P.; Nzoughet, J.K.; Lenaers, G.; Andres, C.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Gelpi, E.; Jové, M.; Mota-Martorell, N.; Obis, È.; Portero-Otin, M.; Povedano, M.; Pujol, A.; Pamplona, R.; Ferrer, I. Lipid alterations in human frontal cortex in ALS-FTLD-TDP43 proteinopathy spectrum are partly related to peroxisome impairment. Neuropathol. Appl. Neurobiol. 2020, 47, 544–563. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Highton-Williamson, E.; Caga, J.; Thornton, N.; Ramsey, E.; Zoing, M.; Kim, W.S.; Halliday, G.M.; Piguet, O.; Hodges, J.R.; et al. Lipid Metabolism and Survival Across the Frontotemporal Dementia-Amyotrophic Lateral Sclerosis Spectrum: Relationships to Eating Behavior and Cognition. J. Alzheimer’s Dis. 2017, 61, 773–783. [Google Scholar] [CrossRef]
- Kim, W.S.; He, Y.; Phan, K.; Ahmed, R.M.; Rye, K.-A.; Piguet, O.; Hodges, J.R.; Halliday, G.M. Altered High Density Lipoprotein Composition in Behavioral Variant Frontotemporal Dementia. Front. Neurosci. 2018, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, H.; Wang, Y.; Zhang, M.; Zhou, Y. Plasma cholesterol in Alzheimer’s disease and frontotemporal dementia. Transl. Neurosci. 2020, 11, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Jary, E.; Pickford, R.; He, Y.; Ahmed, R.M.; Piguet, O.; Hodges, J.R.; Halliday, G.M. Lipidomics Analysis of Behavioral Variant Frontotemporal Dementia: A Scope for Biomarker Development. Front. Neurol. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, K.; He, Y.; Pickford, R.; Bhatia, S.; Katzeff, J.S.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Uncovering pathophysiological changes in frontotemporal dementia using serum lipids. Sci. Rep. 2020, 10, 3640. [Google Scholar] [CrossRef]
- He, Y.; Phan, K.; Bhatia, S.; Pickford, R.; Fu, Y.; Yang, Y.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Increased VLCFA-lipids and ELOVL4 underlie neurodegeneration in frontotemporal dementia. Sci. Rep. 2021, 11, 21348. [Google Scholar] [CrossRef]
- Chen, X.; Yazdani, S.; Piehl, F.; Magnusson, P.K.; Fang, F. Polygenic link between blood lipids and amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 67, 202.e1–202.e6. [Google Scholar] [CrossRef] [Green Version]
- Bandres-Ciga, S.; Noyce, A.J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; Arosio, A.; Barberis, M.; Bartolomei, I.; Battistini, S.; et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol. 2019, 85, 470–481. [Google Scholar] [CrossRef]
- Esteban-García, N.; Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Ayala, J.L.; Matias-Guiu, J.A.; Corrochano, S. Body Complexion and Circulating Lipids in the Risk of TDP-43 Related Disorders. Front. Aging Neurosci. 2022, 14, 838141. [Google Scholar] [CrossRef]
- Mariosa, D.; Hammar, N.; Malmström, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef]
- Iacoangeli, A.; Lin, T.; Al Khleifat, A.; Jones, A.R.; Opie-Martin, S.; Coleman, J.R.; Shatunov, A.; Sproviero, W.; Williams, K.L.; Garton, F.; et al. Genome-wide Meta-analysis Finds the ACSL5-ZDHHC6 Locus Is Associated with ALS and Links Weight Loss to the Disease Genetics. Cell Rep. 2020, 33, 108323. [Google Scholar] [CrossRef]
- Adamo, K.; Dent, R.; Langefeld, C.D.; Cox, M.; Williams, K.; Carrick, K.M.; Stuart, J.S.; Sundseth, S.S.; Harper, M.-E.; McPherson, R.; et al. Peroxisome Proliferator-activated Receptor γ 2 and Acyl-CoA Synthetase 5 Polymorphisms Influence Diet Response. Obesity 2007, 15, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, L.; Huang, T.; Fan, D. Life Course Adiposity and Amyotrophic Lateral Sclerosis: A Mendelian Randomization Study. Ann. Neurol. 2020, 87, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.; Dekker, A.M.; Westeneng, H.-J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Landin-Romero, R.; Liang, C.T.; Keogh, J.M.; Henning, E.; Strikwerda-Brown, C.; Devenney, E.M.; Hodges, J.R.; Kiernan, M.C.; Farooqi, S.; et al. Neural networks associated with body composition in frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 1707–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.M.; MacMillan, M.; Bartley, L.; Halliday, G.M.; Kiernan, M.C.; Hodges, J.R.; Piguet, O. Systemic metabolism in frontotemporal dementia. Neurology 2014, 83, 1812–1818. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Mioshi, E.; Caga, J.; Shibata, M.; Zoing, M.; Bartley, L.; Piguet, O.; Hodges, J.R.; Kiernan, M.C. Body mass index delineates ALS from FTD: Implications for metabolic health. J. Neurol. 2014, 261, 1774–1780. [Google Scholar] [CrossRef]
- Eid, H.R.; Rosness, T.A.; Bosnes, O.; Salvesen, Ø.; Knutli, M.; Stordal, E. Smoking and Obesity as Risk Factors in Frontotemporal Dementia and Alzheimer’s Disease: The HUNT Study. Dement. Geriatr. Cogn. Disord. Extra 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Golimstok, A.; Cámpora, N.; Rojas, J.I.; Fernandez, M.C.; Elizondo, C.M.; Soriano, E.; Cristiano, E. Cardiovascular risk factors and frontotemporal dementia: A case–control study. Transl. Neurodegener. 2014, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Reiser, G. Brain Lipotoxicity of Phytanic Acid and Very Long-chain Fatty Acids. Harmful Cellular/Mitochondrial Activities in Refsum Disease and X-Linked Adrenoleukodystrophy. Aging Dis. 2016, 7, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Hopiavuori, B.R.; Deák, F.; Wilkerson, J.L.; Brush, R.S.; Rocha-Hopiavuori, N.A.; Hopiavuori, A.R.; Ozan, K.G.; Sullivan, M.T.; Wren, J.D.; Georgescu, C.; et al. Homozygous Expression of Mutant ELOVL4 Leads to Seizures and Death in a Novel Animal Model of Very Long-Chain Fatty Acid Deficiency. Mol. Neurobiol. 2017, 55, 1795–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akyol, S.; Ugur, Z.; Yilmaz, A. Lipid profiling of Alzheimer’s disease brain highlights enrichment in glycer-ol(Phospho)lipid, and sphingolipid metabolism. Cells 2021, 10, 2591. [Google Scholar] [CrossRef] [PubMed]
- Serum Lipid Alterations in GBA—Associated Parkinson’s Disease—ClinicalKey. Available online: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S135380201730322X?returnurl=https:%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS135380201730322X%3Fshowall%3Dtrue&referrer=https:%2F%2Fpubmed.ncbi.nlm.nih.gov%2F (accessed on 29 April 2022).
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Ferrari, R.; Heutink, P.; Hardy, J.; Pijnenburg, Y.; Posthuma, D. Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 2017, 140, 1437–1446. [Google Scholar] [CrossRef]
- Su, W.-H.; Shi, Z.-H.; Liu, S.-L.; Wang, X.-D.; Liu, S.; Ji, Y. Updated meta-analysis of the role of APOE ε2/ε3/ε4 alleles in frontotemporal lobar degeneration. Oncotarget 2017, 8, 43721–43732. [Google Scholar] [CrossRef] [Green Version]
- Hertzel, A.V.; Thompson, B.R.; Wiczer, B.M.; Bernlohr, D.A. Lipid metabolism in adipose tissue. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Morris, M.C.; Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol. Aging 2014, 35, S59–S64. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, H.; Ho, W.Y.; Chang, J.; Ling, S. Cholesterol dyshomeostasis in amyotrophic lateral sclerosis: Cause, consequence, or epiphenomenon? FEBS J. 2021. [Google Scholar] [CrossRef]
- Huang, X.; Chen, H.; Miller, W.C.; Mailman, R.B.; Woodard, J.L.; Chen, P.C.; Xiang, D.; Murrow, R.W.; Wang, Y.-Z.; Poole, C. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov. Disord. 2007, 22, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A systematic review and meta-analysis of serum cholesterol and triglyceride levels in patients with Parkinson’s disease. Lipids Health Dis. 2020, 19, 97. [Google Scholar] [CrossRef]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the Cholesterol Biosynthetic Pathway in Huntington’s Disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar] [CrossRef]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Sodero, A.O. 24S-hydroxycholesterol: Cellular effects and variations in brain diseases. J. Neurochem. 2020, 157, 899–918. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Vlad, B.; Gaur, N.; Stubendorff, B.; Grosskreutz, J. Blood–Brain Barrier Disruption Is Not Associated With Disease Aggressiveness in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 1438. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
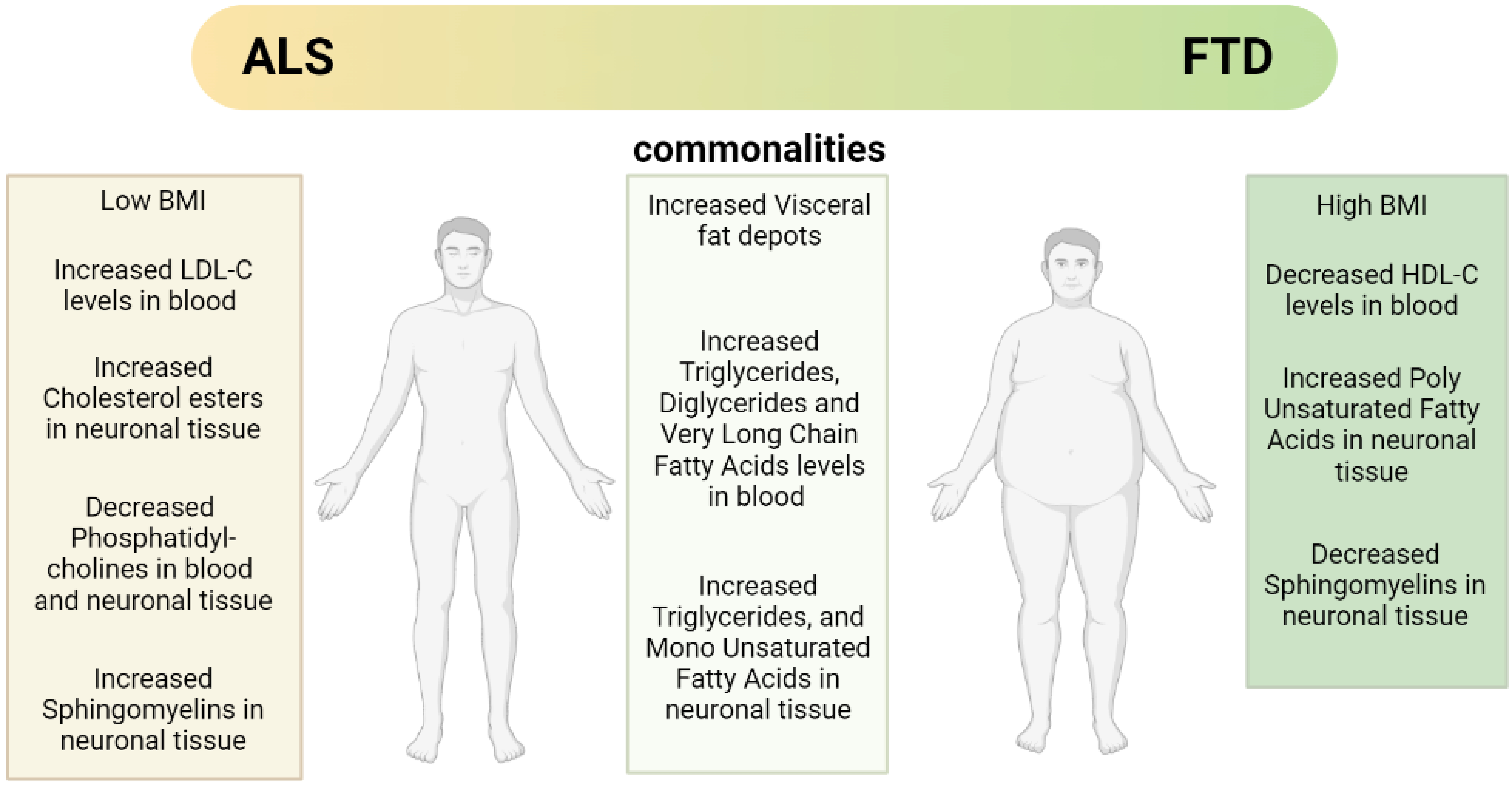
| Lipid Class | Tissue | ALS | FTD |
|---|---|---|---|
| Sterols Lipids | Blood | Up: LDL, LDL/HDL, 25-OHC Down: 27-OHC Refs. [52,53,54,65,67] | Up: Down: HDL Refs. [90,91] |
| CSF | Up: 25-OHC Down: TC, 24-OHC Refs. [59,65] | n.d. | |
| Neuronal Tissue | Up: CEs, 24-OHC, 7α-OHC Down: Refs. [60,61,66] | Not significant changes found Ref. [89] | |
| Fatty Acyls | Blood | Up: TGs, DGs, VLCFA Down: MGs Refs. [53,56,74,75,77,78] | Up: TGs, DGs, VLCFAs Down: Refs. [91,92,93,94,95] |
| CSF | Up: Down: TGs Refs. [79,88] | n.d. | |
| Neuronal Tissue | Up: MUFAs, TGs Down: PUFAs, SFAs Refs. [61,81] | Up: TGs, MUFAs, PUFAs Down: Refs. [92,94] | |
| Sphingolipids | Blood | Up: Cer, GlycoSph Down: SMs Refs. [56,78,79] | Not significant changes found Ref. [93] |
| CSF | Up: GlycoSph, SMs Down: Refs. [79,86,88] | n.d. | |
| Neuronal Tissue | Up: Cer, GlycoSph, SMs Down: Refs. [60,85] | Up: Down: SMs Ref. [89] | |
| Glycerophospholipids | Blood | Up: PE Down: PC, PCe, PS, Pep Refs. [56,78] | Up: Down: PS, PG Refs. [93,94] |
| CSF | Up: PC Down: Refs. [79,88] | n.d. | |
| Neuronal Tissue | Up: Down: PC Ref. [89] | Up: Down: PC, PE Ref. [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godoy-Corchuelo, J.M.; Fernández-Beltrán, L.C.; Ali, Z.; Gil-Moreno, M.J.; López-Carbonero, J.I.; Guerrero-Sola, A.; Larrad-Sainz, A.; Matias-Guiu, J.; Matias-Guiu, J.A.; Cunningham, T.J.; et al. Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders. Biomedicines 2022, 10, 1105. https://doi.org/10.3390/biomedicines10051105
Godoy-Corchuelo JM, Fernández-Beltrán LC, Ali Z, Gil-Moreno MJ, López-Carbonero JI, Guerrero-Sola A, Larrad-Sainz A, Matias-Guiu J, Matias-Guiu JA, Cunningham TJ, et al. Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders. Biomedicines. 2022; 10(5):1105. https://doi.org/10.3390/biomedicines10051105
Chicago/Turabian StyleGodoy-Corchuelo, Juan Miguel, Luis C. Fernández-Beltrán, Zeinab Ali, María J. Gil-Moreno, Juan I. López-Carbonero, Antonio Guerrero-Sola, Angélica Larrad-Sainz, Jorge Matias-Guiu, Jordi A. Matias-Guiu, Thomas J. Cunningham, and et al. 2022. "Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders" Biomedicines 10, no. 5: 1105. https://doi.org/10.3390/biomedicines10051105
APA StyleGodoy-Corchuelo, J. M., Fernández-Beltrán, L. C., Ali, Z., Gil-Moreno, M. J., López-Carbonero, J. I., Guerrero-Sola, A., Larrad-Sainz, A., Matias-Guiu, J., Matias-Guiu, J. A., Cunningham, T. J., & Corrochano, S. (2022). Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders. Biomedicines, 10(5), 1105. https://doi.org/10.3390/biomedicines10051105