
Functional Enrichment Analysis of Regulatory Elements

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.1.1. Gene/Protein and Regulatory Elements Data Collection
2.1.2. Annotation Data Collection
2.2. Analysis Workflow and Results Interpretation
2.2.1. Co-Annotations Discovery Algorithm
2.2.2. Statistics Methods
2.2.3. Handling the Gene Selection Bias
2.2.4. Results Visualisations
2.3. Web Application Implementation
3. Results
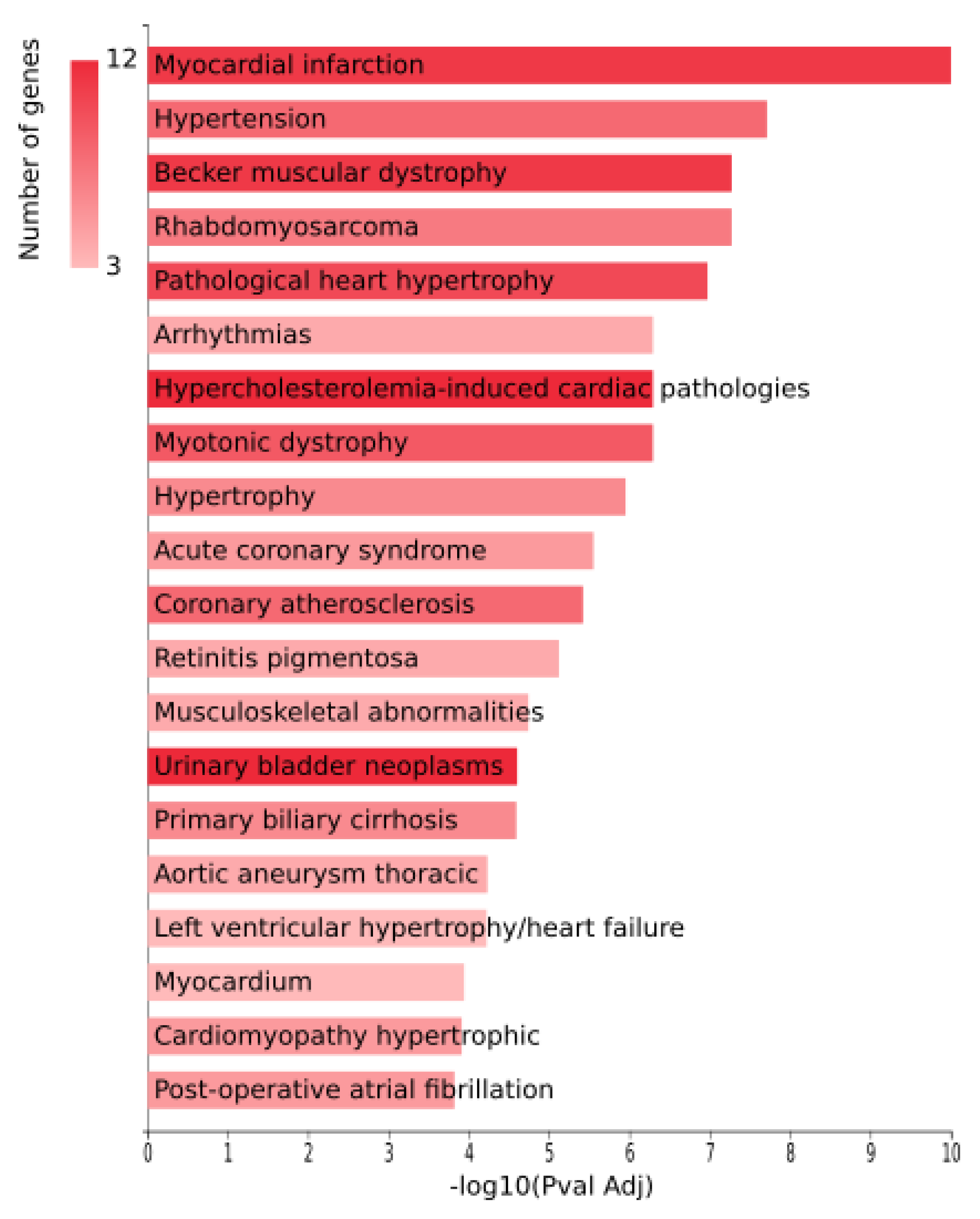
3.1. Analysis of Arrhythmia Related miRNAs
- (a)
- Wallenius test of the miRNAs target genes
- (b)
- Hypergeometric standard test of the transformed miRNAs-based annotations
- (c)
- Hypergeometric standard test of the direct miRNAs-based annotations
- (d)
- Hypergeometric standard test of miRNAs target genes
3.2. Integration in a Complete miRNAs-Seq Analysis Suite
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, A. Gene Set Analysis Methods for the Functional Interpretation of Non-MRNA Data—Genomic Range and NcRNA Data. Brief. Bioinform. 2020, 21, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Tipney, H.; Hunter, L. An Introduction to Effective Use of Enrichment Analysis Software. Hum. Genom. 2010, 4, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Jauhari, S.; Mora, A. Popularity and Performance of Bioinformatics Software: The Case of Gene Set Analysis. BMC Bioinform. 2021, 22, 191. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Mitrea, C.; Taghavi, Z.; Bokanizad, B.; Hanoudi, S.; Tagett, R.; Donato, M.; Voichiţa, C.; Drăghici, S. Methods and Approaches in the Topology-Based Analysis of Biological Pathways. Front. Physiol. 2013, 4, 278. [Google Scholar] [CrossRef] [Green Version]
- Mathur, R.; Rotroff, D.; Ma, J.; Shojaie, A.; Motsinger-Reif, A. Gene Set Analysis Methods: A Systematic Comparison. BioData Min. 2018, 11, 8. [Google Scholar] [CrossRef]
- Geistlinger, L.; Csaba, G.; Santarelli, M.; Ramos, M.; Schiffer, L.; Turaga, N.; Law, C.; Davis, S.; Carey, V.; Morgan, M.; et al. Toward a Gold Standard for Benchmarking Gene Set Enrichment Analysis. Brief. Bioinform. 2021, 22, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Magnusson, R.; Lubovac-Pilav, Z. TFTenricher: A Python Toolbox for Annotation Enrichment Analysis of Transcription Factor Target Genes. BMC Bioinform. 2021, 22, 440. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kuan, P.F. MethylGSA: A Bioconductor Package and Shiny App for DNA Methylation Data Length Bias Adjustment in Gene Set Testing. Bioinforma. Oxf. Engl. 2019, 35, 1958–1959. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Fehlmann, T.; Solomon, J.; Schwed, L.; Grammes, N.; Backes, C.; Van Keuren-Jensen, K.; Craig, D.W.; Meese, E.; Keller, A. MiEAA 2.0: Integrating Multi-Species MicroRNA Enrichment Analysis and Workflow Management Systems. Nucleic Acids Res. 2020, 48, W521–W528. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Wakefield, M.J. Transcript Length Bias in RNA-Seq Data Confounds Systems Biology. Biol. Direct 2009, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Hartnett, L.; Egan, L.J.; Golden, A.; Raja Ali, R.A.; Seoighe, C. Gene-Set Analysis Is Severely Biased When Applied to Genome-Wide Methylation Data. Bioinforma. Oxf. Engl. 2013, 29, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Godard, P.; van Eyll, J. Pathway Analysis from Lists of MicroRNAs: Common Pitfalls and Alternative Strategy. Nucleic Acids Res. 2015, 43, 3490–3497. [Google Scholar] [CrossRef]
- Bleazard, T.; Lamb, J.A.; Griffiths-Jones, S. Bias in MicroRNA Functional Enrichment Analysis. Bioinforma. Oxf. Engl. 2015, 31, 1592–1598. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Saez, P.; Chagoyen, M.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GENECODIS: A Web-Based Tool for Finding Significant Concurrent Annotations in Gene Lists. Genome Biol. 2007, 8, R3. [Google Scholar] [CrossRef] [Green Version]
- Nogales-Cadenas, R.; Carmona-Saez, P.; Vazquez, M.; Vicente, C.; Yang, X.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GeneCodis: Interpreting Gene Lists through Enrichment Analysis and Integration of Diverse Biological Information. Nucleic Acids Res. 2009, 37, W317–W322. [Google Scholar] [CrossRef] [Green Version]
- Tabas-Madrid, D.; Nogales-Cadenas, R.; Pascual-Montano, A. GeneCodis3: A Non-Redundant and Modular Enrichment Analysis Tool for Functional Genomics. Nucleic Acids Res. 2012, 40, W478–W483. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2019, gkz896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alonso, L.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and Integration of Resources for the Estimation of Human Transcription Factor Activities. bioRxiv 2018, 337915. [Google Scholar] [CrossRef]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E. Mouse Genome Database Group Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; A Miller, R.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting Communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA Set Analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A Database for Experimentally Supported Human MicroRNA–Disease Associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal NcRNA–Disease Repository with Increased Coverage and Annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef] [PubMed]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next Generation Access Point for Perturbation-Response Signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The Pharmacogenomics Knowledge Base. In Pharmacogenomics; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1015, pp. 311–320. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) Knowledge Base and Resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, R.; Srikant, R. Fast Algorithms for Mining Association Rules. In Proceedings of the 20th International Conference on Very Large Data Bases, Santiago de Chile, Chile, 12–15 September 1994; Volume 1215. [Google Scholar]
- Wu, F.; Luo, K.; Yan, Z.; Zhang, D.; Yan, Q.; Zhang, Y.; Yi, X.; Zhang, J. Analysis of MiRNAs and Their Target Genes in Five Melilotus Albus NILs with Different Coumarin Content. Sci. Rep. 2018, 8, 14138. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Oshlack, A.; Phipson, B. Gene Set Enrichment Analysis for Genome-Wide DNA Methylation Data. Genome Biol. 2021, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinforma. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Raschka, S. MLxtend: Providing Machine Learning and Data Science Utilities and Extensions to Python’s Scientific Computing Stack. J. Open Source Softw. 2018, 3, 638. [Google Scholar] [CrossRef]
- Servén, D.; Brummitt, C.; Abedi, H. hlink Dswah/PyGAM: V0.8.0; Zenodo; European Organization for Nuclear Research: Geneva, Switzerland, 2018. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H. MicroRNA Regulation of Cardiac Conduction and Arrhythmias. Transl. Res. J. Lab. Clin. Med. 2013, 161, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. SRNAbench and SRNAtoolbox 2019: Intuitive Fast Small RNA Profiling and Differential Expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Approaches | GO BP | PharmGKB | HPO | MNDR |
|---|---|---|---|---|
| Wallenius target-genes | 7 | 9 | 5 | - |
| Transformed DBs | 20 | 19 | 17 | - |
| miRNAs-based DBs | - | - | - | 15 |
| Hypergeometric target-genes | 4 | 6 | 8 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Moreno, A.; López-Domínguez, R.; Villatoro-García, J.A.; Ramirez-Mena, A.; Aparicio-Puerta, E.; Hackenberg, M.; Pascual-Montano, A.; Carmona-Saez, P. Functional Enrichment Analysis of Regulatory Elements. Biomedicines 2022, 10, 590. https://doi.org/10.3390/biomedicines10030590
Garcia-Moreno A, López-Domínguez R, Villatoro-García JA, Ramirez-Mena A, Aparicio-Puerta E, Hackenberg M, Pascual-Montano A, Carmona-Saez P. Functional Enrichment Analysis of Regulatory Elements. Biomedicines. 2022; 10(3):590. https://doi.org/10.3390/biomedicines10030590
Chicago/Turabian StyleGarcia-Moreno, Adrian, Raul López-Domínguez, Juan Antonio Villatoro-García, Alberto Ramirez-Mena, Ernesto Aparicio-Puerta, Michael Hackenberg, Alberto Pascual-Montano, and Pedro Carmona-Saez. 2022. "Functional Enrichment Analysis of Regulatory Elements" Biomedicines 10, no. 3: 590. https://doi.org/10.3390/biomedicines10030590
APA StyleGarcia-Moreno, A., López-Domínguez, R., Villatoro-García, J. A., Ramirez-Mena, A., Aparicio-Puerta, E., Hackenberg, M., Pascual-Montano, A., & Carmona-Saez, P. (2022). Functional Enrichment Analysis of Regulatory Elements. Biomedicines, 10(3), 590. https://doi.org/10.3390/biomedicines10030590