
Tau mRNA Metabolism in Neurodegenerative Diseases: A Tangle Journey



{kind=link}
{kind=link}
Abstract
:1. Tau Protein and Associated Pathologies
2. Tau mRNA Metabolism
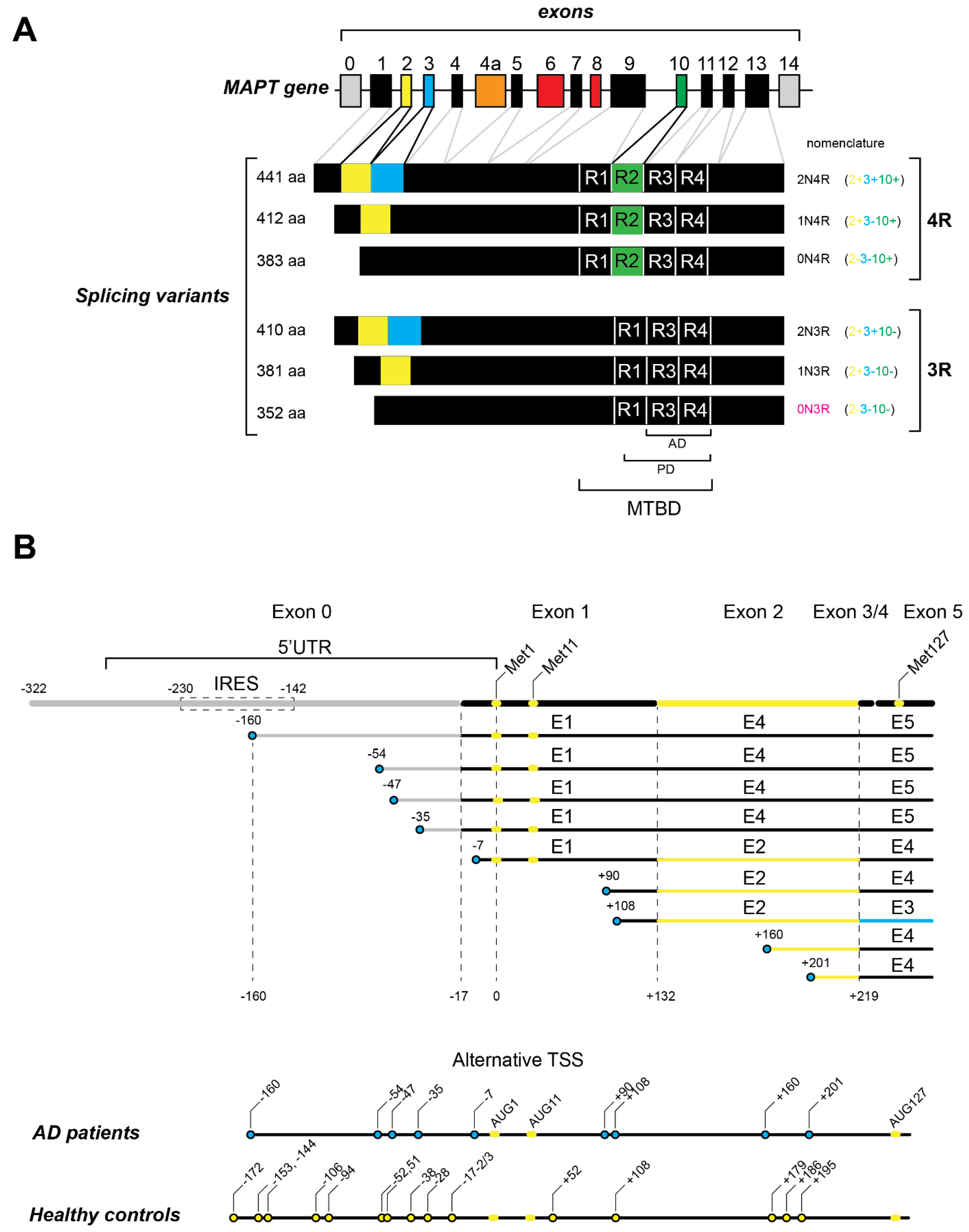
2.1. Splicing Variants
2.2. Tau Transcription and Variable 5′ UTR
2.3. The 3′ UTR Is Important for Tau mRNA Trafficking
2.4. Noncoding RNAs: From microRNAs to Long Noncoding RNAs
3. Tau Protein Metabolism
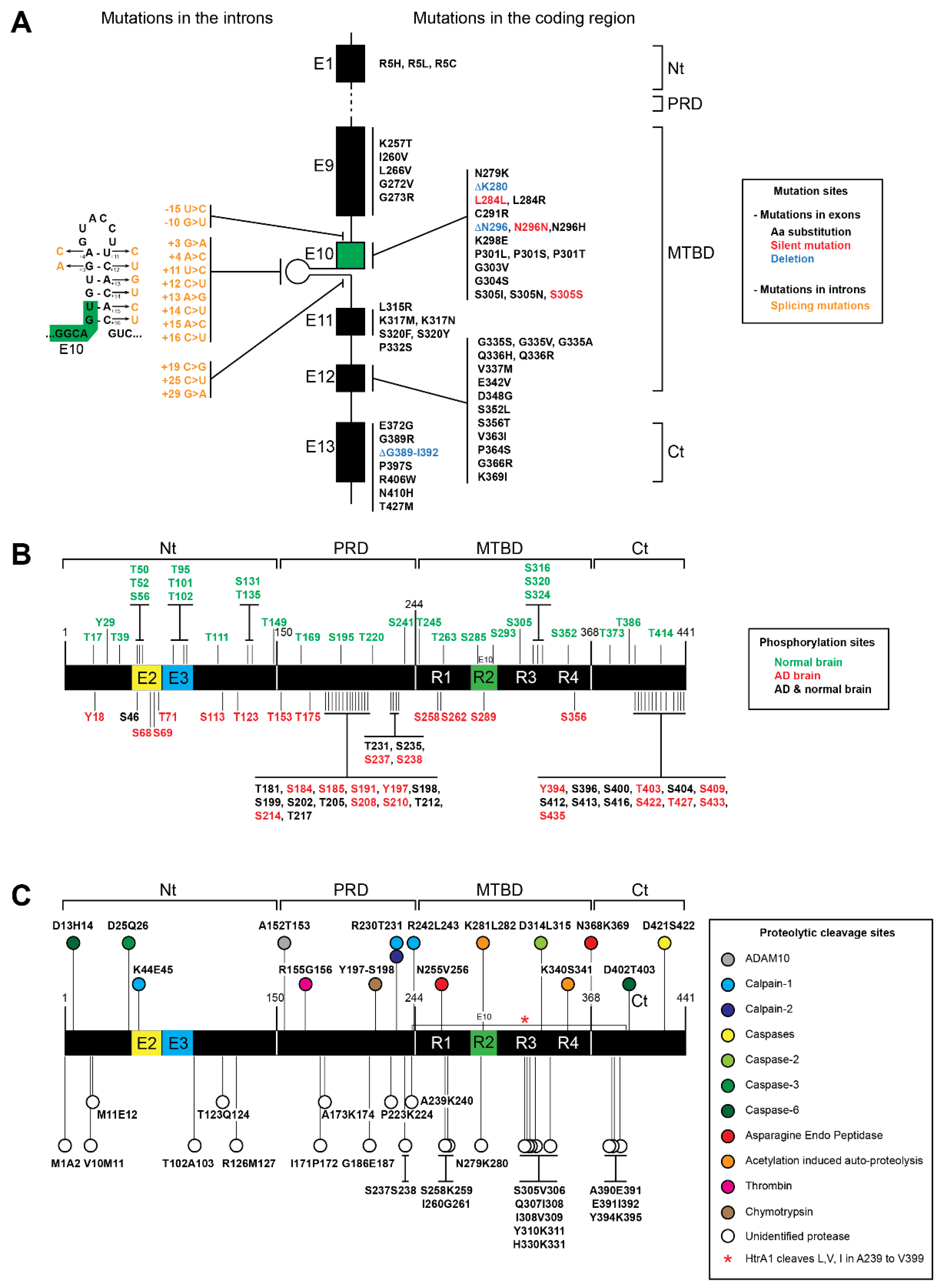
3.1. MAPT Mutations of Tau Proteins
3.2. Post-Translational Modifications of Tau Protein
3.3. Tau Proteolysis
4. Tau Protein and Cellular mRNA Metabolism
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleveland, D.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous central nervous. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Crowther, R.A.; Garner, C.C. Molecular characterization of microtubule-associated proteins tau and map2. Trends Neurosci. 1991, 14, 193–199. [Google Scholar] [CrossRef]
- Janning, D.; Igaev, M.; Sündermann, F.; Brühmann, J.; Beutel, O.; Heinisch, J.J.; Bakota, L.; Piehler, J.; Junge, W.; Brandt, R. Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell 2014, 25, 3541–3551. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, I.; Galas, M.-C.; Silva, J.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.-M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef]
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Chouala, M.S.; Sultan, A.; Marciniak, E.; Humez, S.; Binder, L.; Kayed, R.; et al. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis. 2015, 82, 540–551. [Google Scholar] [CrossRef]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.-C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Lei, S.; Shattuck, J.; Boudeau, S.; Carlomagno, Y.; Medalla, M.; Mashimo, B.L.; Socorro, G.; Al-Mohanna, L.F.A.; Jiang, L.; et al. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; Suchowerska, A.; Van Der Hoven, J.; De Silva, D.M.; Wu, C.W.; van Eersel, J.; Ittner, A.; Ittner, L. Lessons from Tau-Deficient Mice. Int. J. Alzheimer’s Dis. 2012, 2012, 873270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Whitcomb, D.; Eunjoon, K.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. B: Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [Green Version]
- Biundo, F.; Del Prete, D.; Zhang, H.; Arancio, O.; D’Adamio, L. A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 2018, 8, 3184. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Trushina, N.I.; Bakota, L. Much More Than a Cytoskeletal Protein: Physiological and Pathological Functions of the Non-microtubule Binding Region of Tau. Front. Neurol. 2020, 11, 590059. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Ordered Assembly of Tau Protein and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1184, 3–21. [Google Scholar]
- Sexton, C.; Snyder, H.; Beher, D.; Boxer, A.L.; Brannelly, P.; Brion, J.P.; Buée, L.; Cacace, A.M.; Chételat, G.; Citron, M. Current directions in tau research: Highlights from Tau. Alzheimer’s Dement. 2021, 1–20. [Google Scholar] [CrossRef]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef] [Green Version]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Khurana, V.; Merlo, P.; DuBoff, B.; Fulga, T.A.; Sharp, K.A.; Campbell, S.D.; Götz, J.; Feany, M.B. A neuroprotective role for the DNA damage checkpoint in tauopathy. Aging Cell 2012, 11, 360–362. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelison, G.L.; Levy, S.A.; Jenson, T.; Frost, B. Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell 2018, 18, e12847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.-C.; Guo, C.; Yalamanchili, H.K.; Abreha, M.; Al-Ouran, R.; Li, Y.; Dammer, E.B.; Lah, J.J.; Levey, A.I.; Bennett, D.A.; et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 2019, 29, 301–316.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bou Samra, E.; Buhagiar-Labarchède, G.; Machon, C.; Guitton, J.; Onclercq-Delic, R.; Green, M.R.; Alibert, O.; Gazin, C.; Veaute, X.; Amor-Guéret, M. A role for Tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat. Commun. 2017, 8, 693. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Bell, M.; Lyons, D.N.; Rodriguez-Rivera, J.; Ingram, A.; Fontaine, S.N.; Mechas, E.; Chen, J.; Wolozin, B.; LeVine, H.; et al. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J. Neurosci. 2016, 36, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef] [Green Version]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 1990, 9, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Song, X.; Nisbet, R.; Götz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J. Biol. Chem. 2016, 291, 8173–8188. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Congdon, E.; Nagaraja, H.N.; Kuret, J. Tau Isoform Composition Influences Rate and Extent of Filament Formation. J. Biol. Chem. 2012, 287, 20711–20719. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, T.M.; Joachim, C.; Wade-Martins, R. Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol. Aging 2008, 29, 1923–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Kosik, K.S. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol. Cell 2001, 12, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Goode, B.L.; Chau, M.; Denis, P.E.; Feinstein, S.C. Structural and functional differences between 3-repeat and 4-repeat tau isoforms: Implications for normal tau function and the onset of neurodegenerative disease. J. Biol. Chem. 2000, 275, 38182–38189. [Google Scholar] [CrossRef] [Green Version]
- Buée, L.; Delacourte, A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degenera-tion, FTDP-17 and Pick’s disease. Brain Pathol. 1999, 9, 681–693. [Google Scholar] [CrossRef]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Microtubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 643115. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; de Haro, M.; Hao, S.; Park, J.; Rousseaux, M.W.C.; Al-Ramahi, I.; Jafar-Nejad, P.; Vilanova-Velez, L.; See, L.; De Maio, A.; et al. Reduction of Nuak Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 2016, 92, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.; Taylor, M.; Engström, P.; Frith, M.; et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Golovine, K.; Schwerin, M.; Vanselow, J. Three Different Promoters Control Expression of the Aromatase Cytochrome PGene (Cyp19) in Mouse Gonads and Brain1. Biol. Reprod. 2003, 68, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.M.; Rizzu, P.; Francescatto, M.; Vitezic, M.; Leday, G.G.; Sanchez, J.S.; Khamis, A.; Takahashi, H.; van de Berg, W.D.; Medvedeva, Y.A.; et al. Regional differences in gene expression and promoter usage in aged human brains. Neurobiol. Aging 2013, 34, 1825–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.H.; Liu, T.Y.C.; Li, H.; Choo, K.B. Use of a common promoter by two juxtaposed and intronless mouse early embryonic genes, Rnf33 and Rnf35: Implications in zygotic gene expression. Genomics 2002, 80, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Makrigiannis, A.P.; Hodge, D.L.; Anderson, S.K. Identification of a novel Ly49 promoter that is active in bone marrow and fetal thymus. J. Immunol. 2002, 168, 5163–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillet-Boudin, M.-L.; Buee, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Huin, V.; Buée, L.; Behal, H.; Labreuche, J.; Sablonniere, B.; Dhaenens, C.-M. Alternative promoter usage generates novel shorter MAPT mRNA transcripts in Alzheimer’s disease and progressive supranuclear palsy brains. Sci. Rep. 2017, 7, 12589. [Google Scholar] [CrossRef] [Green Version]
- Huin, V.; Deramecourt, V.; Caparros-Lefebvre, D.; Maurage, C.A.; Duyckaerts, C.; Kovari, E.; Pasquier, F.; Buée-Scherrer, V.; Labreuche, J.; Behal, H.; et al. The MAPT gene is differentially methylated in the pro-gressive supranuclear palsy brain. Mov. Disord. 2016, 31, 1883–1890. [Google Scholar] [CrossRef]
- Renbaum, P.; Beeri, R.; Gabai, E.; Amiel, M.; Gal, M.; Ehrengruber, M.U.; Levy-Lahad, E. Egr-1 upregulates the Alzheimer’s disease presenilin-2 gene in neuronal cells. Gene 2003, 318, 113–124. [Google Scholar] [CrossRef]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole Transcriptome Sequencing Reveals Gene Expression and Splicing Differences in Brain Regions Affected by Alzheimer’s Disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef] [PubMed]
- Ounallah-Saad, H.; Beeri, R.; Goshen, I.; Yirmiya, R.; Renbaum, P.; Levy-Lahad, E. Transcriptional regulation of the murine Presenilin-2 gene reveals similarities and differences to its human orthologue. Gene 2009, 446, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Hoffman, B.J. Analysis of a gene encoding two glycine transporter variants reveals alternative promoter usage and a novel gene structure. J. Biol. Chem. 1998, 273, 29077–29085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udono, T.; Yasumoto, K.-I.; Takeda, K.; Amae, S.; Watanabe, K.; Saito, H.; Fuse, N.; Tachibana, M.; Takahashi, K.; Tamai, M.; et al. Structural organization of the human microphthalmia-associated transcription factor gene containing four alternative promoters. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2000, 1491, 205–219. [Google Scholar] [CrossRef]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspa-se-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [Green Version]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Matsumoto, S.-E.; Motoi, Y.; Ishiguro, K.; Tabira, T.; Kametani, F.; Hasegawa, M.; Hattori, N. The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: Implications of prion-like properties. Hum. Mol. Genet. 2015, 24, 6403–6416. [Google Scholar] [CrossRef] [Green Version]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.P.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [Green Version]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buée-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 2015, 5, 9659. [Google Scholar] [CrossRef] [Green Version]
- Veo, B.L.; Krushel, L.A. Translation initiation of the human tau mRNA through an internal ribosomal entry site. J. Alzheimer’s Dis. 2009, 16, 271–275. [Google Scholar] [CrossRef]
- Veo, B.L.; Krushel, L.A. Secondary RNA structure and nucleotide specificity contribute to internal initiation mediated by the human tau 5’ leader. RNA Biol. 2012, 9, 1344–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailliot, J.; Martin, F. Viral internal ribosomal entry sites: Four classes for one goal. Wiley Interdiscip. Rev. RNA 2018, 9, 1–34. [Google Scholar] [CrossRef]
- Lacerda, R.; Menezes, J.; Romão, L. More than just scanning: The importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell Mol. Life Sci. 2016, 74, 1659–1680. [Google Scholar] [CrossRef] [PubMed]
- Logette, E.; Wotawa, A.; Solier, S.; Desoche, L.; Solary, E.; Corcos, L. The human caspase-2 gene: Alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 2003, 22, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Kadener, S.; Fededa, J.P.; Rosbash, M.; Kornblihtt, A. Regulation of alternative splicing by a transcriptional enhancer through RNA pol II elongation. Proc. Natl. Acad. Sci. USA 2002, 99, 8185–8190. [Google Scholar] [CrossRef] [Green Version]
- Cramer, P.; Cáceres, J.F.; Cazalla, D.; Kadener, S.; Muro, A.F.; Baralle, F.E.; Kornblihtt, A.R. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol. Cell 1999, 4, 251–258. [Google Scholar] [CrossRef]
- Aronov, S.L.; Aranda, G.; Behar, L.; Ginzburg, I. Axonal Tau mRNA Localization Coincides with Tau Protein in Living Neuronal Cells and Depends on Axonal Targeting Signal. J. Neurosci. 2001, 21, 6577–6587. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Funakoshi, T.; Sato-Harada, R.; Kanai, Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 1996, 132, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Dennissen, F.J.A.; Kumar, Y.; Luedtke, J.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem. 2017, 292, 12192–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaji, V.; Kaniyappan, S.; Mandelkow, E.; Wang, Y.; Mandelkow, E.M. Pathological missorting of endogenous MAPT/Tau in neurons caused by failure of protein degradation systems. Autophagy 2018, 14, 2139–2154. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Alonso, M.S.; Sündermann, F.; Niewidok, B.; Fernandez, M.-P.; Bakota, L.; Heinisch, J.J.; Brandt, R. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau’s axonal localization. J. Biol. Chem. 2018, 293, 8065–8076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauretti, E.; Dincer, O.; Praticò, D. Regional and temporal miRNAs expression profile in a transgenic mouse model of tauopathy: Implication for its pathogenesis. Mol. Psychiatry 2020, 26, 7020–7028. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. MiR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [Green Version]
- Lekka, E.; Hall, J. Noncoding RNAs in disease. FEBS Lett. 2018, 592, 2884–2900. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Reddy, P.H. MicroRNA-455-3p as a potential biomarker for Alzheimer’s disease: An update. Front. Aging Neurosci. 2018, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Cosín-Tomàs, M.; Antonell, A.; Lladó, A.; Alcolea, D.; Fortea, J.; Ezquerra, M.; Lleó, A.; Martí, M.J.; Pallàs, M.; Sanchez-Valle, R.; et al. Plasma miR-34a-5p and miR-545-3p as Early Biomarkers of Alzheimer’s Disease: Potential and Limitations. Mol. Neurobiol. 2017, 54, 5550–5562. [Google Scholar] [CrossRef] [PubMed]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. Micro RNA -125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Huang, Y.; Wang, L.-L.; Zhang, Y.-F.; Xu, J.; Zhou, Y.; Lourenco, G.; Zhang, B.; Wang, Y.; Ren, R.-J.; et al. MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci. Rep. 2016, 6, 26697. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.-A.; Planel, E.; Sergeant, N.; Buée, L.; Hébert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef] [Green Version]
- Bazrgar, M.; Khodabakhsh, P.; Mohagheghi, F.; Prudencio, M.; Ahmadiani, A. Brain microRNAs dysregulation: Implication for missplicing and abnormal post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Pharmacol. Res. 2020, 155, 104729. [Google Scholar] [CrossRef]
- Praticò, D. The Functional Role of microRNAs in the Pathogenesis of Tauopathy. Cells 2020, 9, 2262. [Google Scholar] [CrossRef]
- Zhao, J.; Yue, D.; Zhou, Y.; Jia, L.; Wang, H.; Guo, M.; Xu, H.; Chen, C.; Zhang, J.; Xu, L. The role of MicroRNAs in Aβ deposition and Tau phosphorylation in Alzheimer’s disease. Front. Neurol. 2017, 8, 342. [Google Scholar] [CrossRef] [Green Version]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Coupland, K.; Kim, W.S.; Halliday, G.; Hallupp, M.; Dobson-Stone, C.; Kwok, J.B.J. Role of the Long Non-Coding RNA MAPT-AS1 in Regulation of Microtubule Associated Protein Tau (MAPT) Expression in Parkinson’s Disease. PLoS ONE 2016, 11, e0157924. [Google Scholar] [CrossRef] [Green Version]
- Speir, M.L.; Zweig, A.S.; Rosenbloom, K.R.; Raney, B.J.; Paten, B.; Nejad, P.; Lee, B.T.; Learned, K.; Karolchik, D.; Hinrichs, A.; et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016, 44, D717–D725. [Google Scholar] [CrossRef]
- Elkouris, M.; Kouroupi, G.; Vourvoukelis, A.; Papagiannakis, N.; Kaltezioti, V.; Matsas, R.; Stefanis, L.; Xilouri, M.; Politis, P.K. Long Non-coding RNAs Associated With Neurodegeneration-Linked Genes Are Reduced in Parkinson’s Disease Patients. Front. Cell. Neurosci. 2019, 13, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, R.; Javad, F.; Emmett, W.; Wilkins, O.G.; Almeida, F.L.; Barahona-Torres, N.; Zareba-Paslawska, J.; Ehteramyan, M.; Zuccotti, P.; Modelska, A.; et al. MIR-NATs repress MAPT translation and aid proteostasis in neurodegeneration. Nature 2021, 594, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Stefanoska, K.; Volkerling, A.; Bertz, J.; Poljak, A.; Ke, Y.; Ittner, L.; Ittner, A. An N-terminal motif unique to primate tau enables differential protein–protein interactions. J. Biol. Chem. 2018, 293, 3710–3719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Ikezu, T. Tau Secretion. Adv. Exp. Med. Biol. 2019, 1184, 123–134. [Google Scholar]
- Hasegawa, M.; Smith, M.J.; Goedert, M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998, 437, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Crowther, R. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999, 450, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Nacharaju, P.; Lewis, J.; Easson, C.; Yen, S.; Hackett, J.; Hutton, M. Accelerated filament formation from Tau protein with SPECIFIC FTDP-17 missense mutations. J. Neuropathol. Exp. Neurol. 1999, 58, 545. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–704. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Yamaguchi, Y.; Mishra, S.K.; Higuchi, M.; Sahara, N. Tau Filaments and the Development of Positron Emission Tomography Tracers. Front. Neurol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2014, 41, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafei, R.; Woollacott, I.O.; Mummery, C.J.; Bocchetta, M.; Guerreiro, R.; Bras, J.; Warren, J.; Lashley, T.; Jaunmuktane, Z.; Rohrer, J.D. Two pathologically confirmed cases of novel mutations in the MAPT gene causing frontotemporal dementia. Neurobiol. Aging 2020, 87, 141.e15–141.e20. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Tagliavini, F. Frontotemporal lobar degeneration: Old knowledge and new insight into the pathogenetic mechanisms of tau mutations. Front. Aging Neurosci. 2015, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gong, C.X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Fairen, M.D.; Sabir, M.S.; Pastor, P.; Ding, J.; Ispierto, L.; Butala, A.; Morris, C.M.; Schulte, C.; Gasser, T.; et al. MAPT p.V363I mutation A rare cause of corticobasal degeneration. Neurol. Genet. 2019, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, I.; Silva, J.; Kimura, T.; Rodrigues, A.J.; Costa, P.; Almeida, O.F.X.; Sousa, N.; Takashima, A. Female hippo-campus vulnerability to environmental stress, a precipitating factor in tau aggregation pathology. J Alzheimer’s Dis. 2015, 43, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Sato, Y.; Takashima, A. Frontotemporal dementia with Parkinsonism linked to chromosome-17 mutations enhance tau oligomer formation. Neurobiol. Aging 2018, 69, 26–32. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Synthetic Tau Fibrils Mediate Transmission of Neurofibrillary Tangles in a Transgenic Mouse Model of Alzheimer’s-Like Tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Donck, L.V.; Torremans, A.; Sluydts, E.; et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.J.; Kerridge, C.; Cooper, J.; Cavallini, A.; Falcon, B.; Cella, C.V.; Landi, A.; Szekeres, P.G.; Murray, T.K.; Ahmed, Z.; et al. Short Fibrils Constitute the Major Species of Seed-Competent Tau in the Brains of Mice Transgenic for Human P301S Tau. J. Neurosci. 2016, 36, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 2408–2421. [Google Scholar] [CrossRef] [Green Version]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.-P.; Hamdane, M.; Buee, L. Alzheimer’s Disease-Like Tau Neuropathology Leads to Memory Deficits and Loss of Functional Synapses in a Novel Mutated Tau Transgenic Mouse without Any Motor Deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Diekmann, S.; Bertsch, U.; Vanmassenhove, B.; Bogerts, B.; Kretzschmar, H.A. Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics 2005, 6, 91–95. [Google Scholar] [CrossRef]
- Ando, K.; Ferlini, L.; Suain, V.; Yilmaz, Z.; Mansour, S.; Le Ber, I.; Bouchard, C.; Leroy, K.; Durr, A.; Clot, F.; et al. De novo MAPT mutation G335A causes severe brain atrophy, 3R and 4R PHF-tau pathology and early onset frontotemporal dementia. Acta Neuropathol. Commun. 2020, 8, 94. [Google Scholar] [CrossRef]
- Origone, P.; Geroldi, A.; Lamp, M.; Sanguineri, F.; Caponnetto, C.; Cabona, C.; Gotta, F.; Trevisan, L.; Bellone, E.; Manganelli, F.; et al. Role of MAPT in Pure Motor Neuron Disease: Report of a Recurrent Mutation in Italian Patients. Neurodegener. Dis. 2019, 18, 310–314. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Ronchi, D.; Gallia, F.; Cribiù, F.M.; Trezzi, I.; Vetro, A.; Mina, E.; Della Limongelli, I.; Bellazzi, R.; Ricca, I.; et al. Lower motor neuron disease with respiratory failure caused by a novel MAPT mutation. Neurology 2014, 82, 1990–1998. [Google Scholar] [CrossRef]
- Momeni, P.; Wickremaratchi, M.M.; Bell, J.; Arnold, R.; Beer, R.; Hardy, J.; Revesz, T.; Neal, J.W.; Morris, H.R. Familial early onset frontotemporal dementia caused by a novel S356T MAPT mutation, initially diagnosed as schizophrenia. Clin. Neurol. Neurosurg. 2010, 112, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Nicholl, D.J.; Greenstone, M.A.; Clarke, C.E.; Rizzu, P.; Crooks, D.; Crowe, A.; Trojanowski, J.Q.; Lee, V.M.-Y.; Heutink, P. An English kindred with a novel recessive tauopathy and respiratory failure. Ann. Neurol. 2003, 54, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Bastone, A.; Piccoli, E.; Mazzoleni, G.; Morbin, M.; Uggetti, A.; Giaccone, G.; Sperber, S.; Beeg, M.; Salmona, M.; et al. New mutations in MAPT gene causing frontotemporal lobar degeneration: Biochemical and structural characteriza-tion. Neurobiol. Aging 2012, 33, 834.e1–834.e6. [Google Scholar]
- Borrego-Écija, S.; Antonell, A.; Puig-Butillé, J.A.; Pericot, I.; Prat-Bravo, C.; Abellan-Vidal, M.T.; Mallada, J.; Olives, J.; Falgàs, N.; Oliva, R.; et al. Novel P397S MAPT variant associated with late onset and slow progressive fronto-temporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 1559–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostojic, J.; Elfgren, C.; Passant, U.; Fabre, S.F.; Nilsson, K.; Gustafson, L.; Lannfelt, L. The Tau R406W Mutation Causes Progressive Presenile Dementia with Bitemporal Atrophy. Dement. Geriatr. Cogn. Disord. 2004, 17, 298–301. [Google Scholar] [CrossRef]
- Giaccone, G.; Rossi, G.; Farina, L.; Marcon, G.; Di Fede, G.; Catania, M.; Morbin, M.; Sacco, L.; Bugiani, O.; Tagliavini, F. Familial frontotemporal dementia associated with the novel MAPT mutation T427M. J. Neurol. 2005, 252, 1543–1545. [Google Scholar] [CrossRef]
- Cruchaga, C.; Chakraverty, S.; Mayo, K.; Vallania, F.L.M.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; Foroud, T.M.; et al. Goate AM & for the NIA-LOAD—NCRAD Family Study Consortium (2012) Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar]
- Poorkaj, P.; Muma, N.A.; Zhukareva, V.; Cochran, E.J.; Shannon, K.M.; Hurtig, H.; Koller, W.C.; Bird, T.D.; Trojanowski, J.Q.; Lee, V.M.-Y.; et al. An R5L τ mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol. 2002, 52, 511–516. [Google Scholar] [CrossRef]
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hull, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Toyoshima, Y.; Hasegawa, M.; Umeda, Y.; Wakabayashi, K.; Tokiguchi, S.; Iwatsubo, T.; Takahashi, H. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann. Neurol. 2002, 51, 525–530. [Google Scholar] [CrossRef]
- Mutreja, Y.; Combs, B.; Gamblin, T.C. FTDP-17 mutations alter the aggregation and microtubule stabilization propensity of tau in an isoform-specific fashion. Biochemistry 2019, 58, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Giovanni, G.D.; et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichou, Y.; Al-Hilaly, Y.K.; Devred, F.; Smet-Nocca, C.; Tsvetkov, P.O.; Verelst, J.; Winderickx, J.; Geukens, N.; Vanmechelen, E.; Perrotin, A.; et al. The elusive tau molecular structures: Can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol. Commun. 2019, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Mair, W.; Muntel, J.; Tepper, K.; Tang, S.; Biernat, J.; Seeley, W.W.; Kosik, K.S.; Mandelkow, E.; Steen, H.; Steen, J.A. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal. Chem. 2016, 88, 3704–3714. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S123–S139. [Google Scholar] [CrossRef]
- Thies, E.; Mandelkow, E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 2007, 27, 2896–2907. [Google Scholar] [CrossRef] [Green Version]
- Boyarko, B.; Hook, V. Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neuro-degeneration. Front. Neurosci. 2021, 15, S123–S139. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; Hascher, A.; Chinnathambi, S.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a patho-logical (MC-1) conformation. J. Biol. Chem. 2008, 283, 32066–32076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malia, T.J.; Teplyakov, A.; Ernst, R.; Wu, S.J.; Lacy, E.R.; Liu, X.; Vandermeeren, M.; Mercken, M.; Luo, J.; Sweet, R.W.; et al. Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins Struct. Funct. Bioinform. 2016, 84, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, P.D.; Tracy, T.E.; Son, H.I.; Zhou, Y.; Leite, R.E.P.; Miller, B.L.; Seeley, W.W.; Grinberg, L.T.; Gan, L. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol. Neurodegener. 2016, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Min, S.-W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.-A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.W.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef]
- Cockram, P.E.; Kist, M.; Prakash, S.; Chen, S.-H.; Wertz, I.E.; Vucic, D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. 2021, 28, 591–605. [Google Scholar] [CrossRef]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The ubiquitin system in AlzheimerTs. Proteostasis Dis. Basic Mech. Clin. 2020, 1233, 195. [Google Scholar]
- Thomas, S.N.; Funk, K.E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A.J. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: A mass spectrometry approach. Acta Neuropathol. 2011, 123, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.P.; Corbett, N.J.; Kellett, K.A.B.; Hooper, N.M. Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis. 2018, 63, 13–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, P.; Cehlar, O.; Skrabana, R.; Novak, M. Tau Conformation as a Target for Disease-Modifying Therapy: The Role of Truncation. J. Alzheimer’s Dis. 2018, 64, S535–S546. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kotilinek, L.A.; Smith, B.; Hlynialuk, C.; Zahs, K.; Ramsden, M.; Cleary, J.; Ashe, K.H. Caspase-2 cleavage of tau reversibly impairs memory. Nat. Med. 2016, 22, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Dinkel, P.D.; Holden, M.R.; Matin, N.; Margittai, M. RNA Binds to Tau Fibrils and Sustains Template-Assisted Growth. Biochemistry 2015, 54, 4731–4740. [Google Scholar] [CrossRef] [Green Version]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Qual. Life Res. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [Green Version]
- Tomé, S.O.; Gomes, L.A.; Li, X.; Vandenberghe, R.; Tousseyn, T.; Thal, D.R. TDP-43 interacts with pathological τ protein in Alzheimer’s disease. Acta Neuropathol. 2021, 141, 795–799. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Maziuk, B.F.; Apicco, D.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apicco, D.; Ash, P.E.A.; Maziuk, B.; LeBlang, C.; Medalla, M.; Al Abdullatif, A.; Ferragud, A.; Botelho, E.; Balance, H.I.; Dhawan, U.; et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 2018, 21, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauderlier, A.; Gilles, M.; Spolcova, A.; Caillierez, R.; Chwastyniak, M.; Kress, M.; Drobecq, H.; Bonnefoy, E.; Pinet, F.; Weil, D.; et al. Tau/DDX6 interaction increases microRNA activity. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1861, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Kedersha, N.; Low, W.-K.; Romo, D.; Gorospe, M.; Kaufman, R.; Anderson, P.; Liu, J.O. Eukaryotic Initiation Factor 2α-independent Pathway of Stress Granule Induction by the Natural Product Pateamine A. J. Biol. Chem. 2006, 281, 32870–32878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic Shuttling of Tia-Accompanies the Recruitment of mRNA to Mammalian Stress Granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence that ternary complex (eIF2-GTP-tRNAiMet)-Deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.-E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of Ribosome Recruitment Induces Stress Granule Formation Independently of Eukaryotic Initiation Factor 2α Phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.-J.; Anderson, P.; Kaufman, R.J. Heme-regulated Inhibitor Kinase-mediated Phosphorylation of Eukaryotic Translation Initiation Factor Inhibits Translation, Induces Stress Granule Formation, and Mediates Survival upon Arsenite Exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [Green Version]
- Mokas, S.; Mills, J.R.; Garreau, C.; Fournier, M.J.; Robert, F.; Arya, P.; Kaufman, R.J.; Pelletier, J.; Mazroui, R. Uncoupling stress granule assembly and translation initiation inhibition. Mol. Biol. Cell 2009, 20, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Parker, R. Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadi, M.R.; Moslehian, M.S.; Sabaie, H.; Jalaiei, A.; Ghafouri-Fard, S.; Taheri, M.; Rezazadeh, M. Stress Granules and Neurodegenerative Disorders: A Scoping Review. Front. Aging Neurosci. 2021, 13, 650740. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ash, P.E.A.; Maziuk, B.F.; Balance, H.I.; Boudeau, S.; Al Abdullatif, A.; Orlando, M.; Petrucelli, L.; Ikezu, T.; Wolozin, B. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 2019, 137, 259–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Rodrigues, S.; Sampaio-Marques, B.; Gomes, P.; Carvalho, A.; Dioli, C.; Soares-Cunha, C.; Mazuik, B.F.; Takashima, A.; Ludovico, P.; et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 2019, 26, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Yu, W.H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting Pathology of the Stress Granule Proteins TIA-1 and G3BP in Tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef] [Green Version]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; da Rocha, E.L.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIARegulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Apicco, D.; Zhang, C.; Maziuk, B.; Jiang, L.; Balance, H.I.; Boudeau, S.; Ung, C.; Li, H.; Wolozin, B. Dysregulation of RNA Splicing in Tauopathies. Cell Rep. 2019, 29, 4377–4388.e4. [Google Scholar] [CrossRef] [Green Version]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McCowan, E.; et al. Medicine: Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ri-bonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005, 33, 6000–6010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 10, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pa-thology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA Binding Protein-43 (TDP-43) Associates with Stress Granules: Analysis of Cultured Cells and Pathological Brain Tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [Green Version]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; Von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Bénard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef]
- Lee, M.; Sadowska, A.; Bekere, I.; Ho, D.; Gully, B.; Lu, Y.; Iyer, K.S.; Trewhella, J.; Fox, A.H.; Bond, C.S. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015, 43, 3826–3840. [Google Scholar] [CrossRef]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef]
- Jiang, L.; Lin, W.; Zhang, C.; Ash, P.E.; Verma, M.; Kwan, J.; van Vliet, E.; Yang, Z.; Cruz, A.L.; Boudeau, S.; et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 2021, 81, 4209–4227.e12. [Google Scholar] [CrossRef]
- Anders, M.; Chelysheva, I.; Goebel, I.; Trenkner, T.; Zhou, J.; Mao, Y.; Verzini, S.; Qian, S.-B.; Ignatova, Z. Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Sci. Alliance 2018, 1, e201800113. [Google Scholar] [CrossRef] [Green Version]
- Arguello, A.E.; DeLiberto, A.N.; Kleiner, R.E. RNA Chemical Proteomics Reveals the N6-Methyladenosine (m6A)-Regulated Protein–RNA Interactome. J. Am. Chem. Soc. 2017, 139, 17249–17252. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Zhang, J.; Zhu, J.-S. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Hamm, M.J.; Meier, S.E.; Weiss, B.E.; Nation, G.K.; Chishti, E.A.; Arango, J.P.; Chen, J.; Zhu, H.; Blalock, E.M.; et al. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019, 137, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.A.; Galvis-Escobar, S.; Abisambra, J.F. Tau-mediated dysregulation of RNA: Evidence for a common molecular mechanism of toxicity in frontotemporal dementia and other tauopathies. Neurobiol. Dis. 2020, 141, 104939. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Puangmalai, N.; Sengupta, U.; Bhatt, N.; Johnson, O.D.; Kharas, M.G.; Kayed, R. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Okano, H.; Blendy, J.A.; Montell, C. Musashi, a neural RNA-binding protein required for drosophila adult external sensory organ development. Neuron 1994, 13, 67–81. [Google Scholar] [CrossRef]
- Kaneko, J.; Chiba, C. Immunohistochemical analysis of Musashi-1 expression during retinal regeneration of adult newt. Neurosci. Lett. 2009, 450, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.; Wang, H.; Schmitt-Ulms, G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell. Proteom. 2015, 14, 3000–3014. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Basquin, J.Ô.; Ozgur, S.; Czarnocki-Cieciura, M.; Bonneau, F.; Aartse, A.; Dziembowski, A.; Nowotny, M.; Conti, E.; Filipowicz, W. Structural and Biochemical Insights to the Role of the CCR4-NOT Complex and DDXATPase in Mi-croRNA Repression. Mol. Cell 2014, 54, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Maina, M.B.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Maina, M.B.; Bailey, L.J.; Doherty, A.J.; Serpell, L.C. The Involvement of Aβ42 and Tau in Nucleolar and Protein Synthesis Machinery Dysfunction. Front. Cell. Neurosci. 2018, 12, 220. [Google Scholar] [CrossRef]
- Cruz, A.; Verma, M.; Wolozin, B. The Pathophysiology of Tau and Stress Granules in Disease. In Tau Biology; Springer: Singapore, 2019; Volume 1184, pp. 359–372. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Costa, P.J.; Hamdane, M.; Buée, L.; Martin, F. Tau mRNA Metabolism in Neurodegenerative Diseases: A Tangle Journey. Biomedicines 2022, 10, 241. https://doi.org/10.3390/biomedicines10020241
da Costa PJ, Hamdane M, Buée L, Martin F. Tau mRNA Metabolism in Neurodegenerative Diseases: A Tangle Journey. Biomedicines. 2022; 10(2):241. https://doi.org/10.3390/biomedicines10020241
Chicago/Turabian Styleda Costa, Paulo J., Malika Hamdane, Luc Buée, and Franck Martin. 2022. "Tau mRNA Metabolism in Neurodegenerative Diseases: A Tangle Journey" Biomedicines 10, no. 2: 241. https://doi.org/10.3390/biomedicines10020241
APA Styleda Costa, P. J., Hamdane, M., Buée, L., & Martin, F. (2022). Tau mRNA Metabolism in Neurodegenerative Diseases: A Tangle Journey. Biomedicines, 10(2), 241. https://doi.org/10.3390/biomedicines10020241