
Expression Profiling in Ovarian Cancer Reveals Coordinated Regulation of BRCA1/2 and Homologous Recombination Genes

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Samples
2.2. RNA isolation and Quantification
2.3. Gene Expression Profiling with the nCounter System
2.4. BRCA1 and BRCA2 mRNA Analysis by Droplet Digital PCR
2.5. Statistical Analysis
2.5.1. General
2.5.2. Analysis of NanoString Data
2.6. The Cancer Genome Atlas (TCGA) Data Analysis
3. Results
3.1. Study Cohort
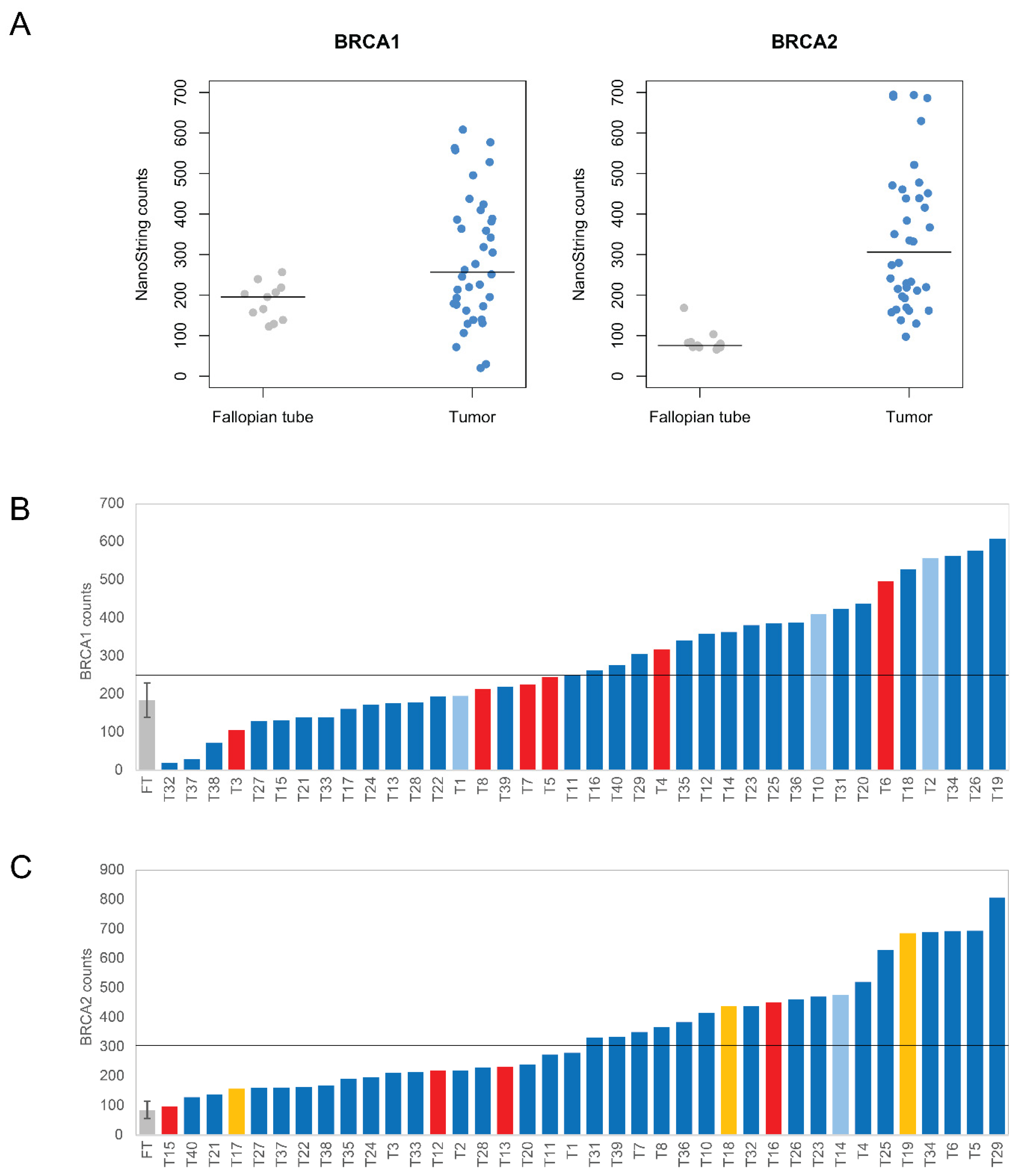
3.2. Quantification of BRCA1 and BRCA2 mRNAs with the nCounter Assay
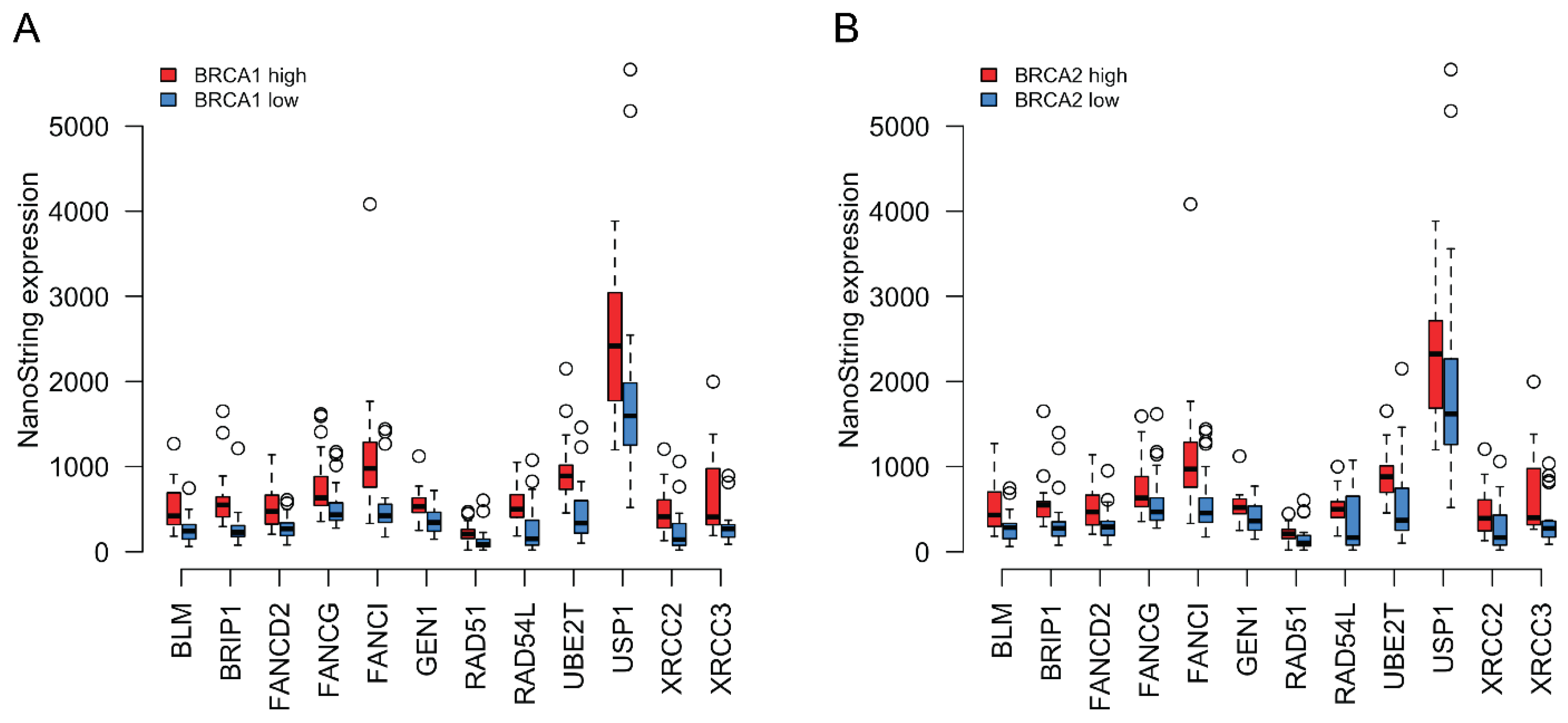
3.3. Expression Profiling of DNA Damage and Repair Genes
3.4. Coordinated Expression of BRCA1/2 and a Subset of HR Genes in TCGA Data
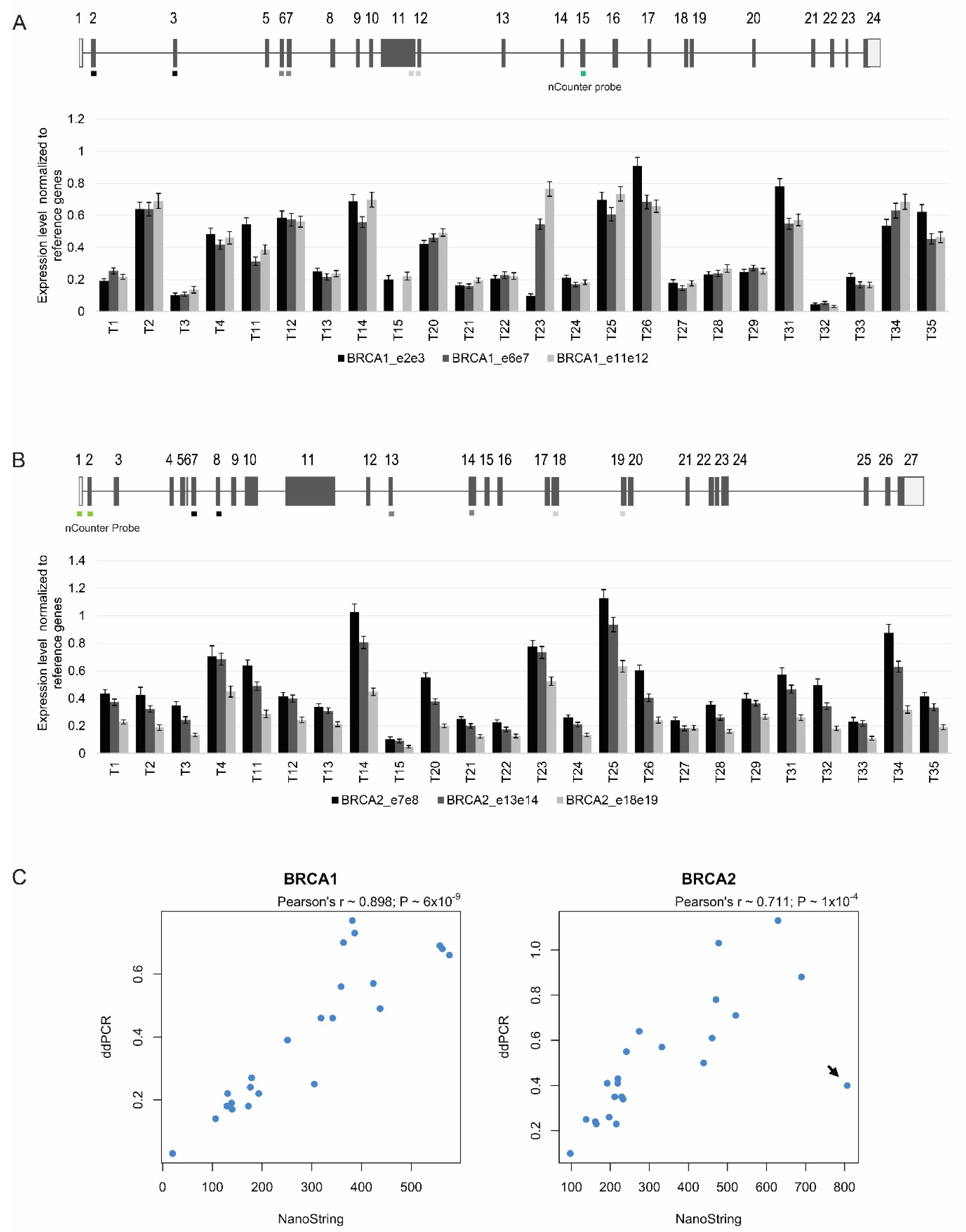
3.5. Targeted Quantitation of BRCA1/2 mRNA by ddPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2016; Based on November 2018 SEER Data Submission; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet. Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef]
- Peng, G.; Chun-Jen Lin, C.; Mo, W.; Dai, H.; Park, Y.Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef]
- Hu, Z.; Yau, C.; Ahmed, A.A. A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat. Commun. 2017, 8, 15943. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- North, B.V.; Curtis, D.; Sham, P.C. A note on calculation of empirical P values from Monte Carlo procedure. Am. J. Hum. Genet. 2003, 72, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Hu, Z.; Artibani, M.; Alsaadi, A.; Wietek, N.; Morotti, M.; Shi, T.; Zhong, Z.; Santana Gonzalez, L.; El-Sahhar, S.; KaramiNejadRanjbar, M.; et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 2020, 37, 226–242.e227. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Masuda, N.; Ohnishi, T.; Kawamoto, S.; Monden, M.; Okubo, K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999, 27, 4436–4443. [Google Scholar] [CrossRef] [PubMed]
- von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA quality from FFPE samples. PLoS ONE 2007, 2, e1261. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef]
- Peixoto, A.; Santos, C.; Pinto, P.; Pinheiro, M.; Rocha, P.; Pinto, C.; Bizarro, S.; Veiga, I.; Principe, A.S.; Maia, S.; et al. The role of targeted BRCA1/BRCA2 mutation analysis in hereditary breast/ovarian cancer families of Portuguese ancestry. Clin. Genet. 2015, 88, 41–48. [Google Scholar] [CrossRef]
- Machado, P.M.; Brandão, R.D.; Cavaco, B.M.; Eugénio, J.; Bento, S.; Nave, M.; Rodrigues, P.; Fernandes, A.; Vaz, F. Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: Evidence for a founder effect and analysis of the associated phenotypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2027–2034. [Google Scholar] [CrossRef]
- Caputo, S.M.; Léone, M.; Damiola, F.; Ehlen, A.; Carreira, A.; Gaidrat, P.; Martins, A.; Brandão, R.D.; Peixoto, A.; Vega, A.; et al. Full in-frame exon 3 skipping of BRCA2 confers high risk of breast and/or ovarian cancer. Oncotarget 2018, 9, 17334–17348. [Google Scholar] [CrossRef]
- Heredia, N.J.; Belgrader, P.; Wang, S.; Koehler, R.; Regan, J.; Cosman, A.M.; Saxonov, S.; Hindson, B.; Tanner, S.C.; Brown, A.S.; et al. Droplet Digital™ PCR quantitation of HER2 expression in FFPE breast cancer samples. Methods 2013, 59, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Meehan, K.; Clynick, B.; Mirzai, B.; Maslen, P.; Harvey, J.M.; Erber, W.N. HER2 mRNA transcript quantitation in breast cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2017, 19, 606–615. [Google Scholar] [CrossRef]
- Gudas, J.M.; Li, T.; Nguyen, H.; Jensen, D.; Rauscher, F.J., 3rd; Cowan, K.H. Cell cycle regulation of BRCA1 messenger RNA in human breast epithelial cells. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1996, 7, 717–723. [Google Scholar]
- Rajan, J.V.; Marquis, S.T.; Gardner, H.P.; Chodosh, L.A. Developmental expression of Brca2 colocalizes with Brca1 and is associated with proliferation and differentiation in multiple tissues. Dev. Biol. 1997, 184, 385–401. [Google Scholar] [CrossRef]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar] [PubMed]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. AKT regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 143. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Wang, L.; Wei, J.; Qian, X.; Yin, H.; Zhao, Y.; Yu, L.; Wang, T.; Liu, B. ERCC1 and BRCA1 mRNA expression levels in metastatic malignant effusions is associated with chemosensitivity to cisplatin and/or docetaxel. BMC Cancer 2008, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.E.; James, C.R.; Stewart, G.E.; Mulligan, J.M.; White, P.; Chang, G.K.; Mullan, P.B.; Johnston, P.G.; Wilson, R.H.; Harkin, D.P. BRCA1 mRNA expression levels predict for overall survival in ovarian cancer after chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 7413–7420. [Google Scholar] [CrossRef]
- Carser, J.E.; Quinn, J.E.; Michie, C.O.; O’Brien, E.J.; McCluggage, W.G.; Maxwell, P.; Lamers, E.; Lioe, T.F.; Williams, A.R.; Kennedy, R.D.; et al. BRCA1 is both a prognostic and predictive biomarker of response to chemotherapy in sporadic epithelial ovarian cancer. Gynecol. Oncol. 2011, 123, 492–498. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, J.; Zhang, X.; Wu, Q.; Jiang, S.; Liu, Y.; Hu, Z.; Liu, B.; Chen, X. BRCA1 mRNA expression as a predictive and prognostic marker in advanced esophageal squamous cell carcinoma treated with cisplatin- or docetaxel-based chemotherapy/chemoradiotherapy. PLoS ONE 2013, 8, e52589. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, P.; Liu, B.; Du, J. Successful personalized chemotherapy for metastatic gastric cancer based on quantitative BRCA1 mRNA expression level: A case report. Oncol. Lett. 2016, 11, 4183–4186. [Google Scholar] [CrossRef][Green Version]
- Rosell, R.; Perez-Roca, L.; Sanchez, J.J.; Cobo, M.; Moran, T.; Chaib, I.; Provencio, M.; Domine, M.; Sala, M.A.; Jimenez, U.; et al. Customized treatment in non-small-cell lung cancer based on EGFR mutations and BRCA1 mRNA expression. PLoS ONE 2009, 4, e5133. [Google Scholar] [CrossRef] [PubMed]
- Taron, M.; Rosell, R.; Felip, E.; Mendez, P.; Souglakos, J.; Ronco, M.S.; Queralt, C.; Majo, J.; Sanchez, J.M.; Sanchez, J.J.; et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum. Mol. Genet. 2004, 13, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Tsibulak, I.; Wieser, V.; Degasper, C.; Shivalingaiah, G.; Wenzel, S.; Sprung, S.; Lax, S.F.; Marth, C.; Fiegl, H.; Zeimet, A.G. BRCA1 and BRCA2 mRNA-expression prove to be of clinical impact in ovarian cancer. Br. J. Cancer 2018, 119, 683–692. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Genes Upregulated in Tumors. | |
| Pathway | Genes |
| Apoptosis | BCL2L1, CASP8, MYD88 |
| Base excision repair | APEX2, FEN1, NEIL3, PARP1, SMUG1, UNG |
| Base excision repair—Translesion Synthesis—Cell Cycle and Signaling | PCNA |
| Cell Cycle and Signaling | AURKA, BUB1B, CDKN2A, KRAS, RAD21, RM12, SUMO3 |
| Checkpoint Activation | H2AFX |
| Checkpoint Activation—Cell Cycle and Signaling | CHEK1/2 |
| Homologous Recombination and Fanconi Anemia | BRIP1, BLM, BRCA2, FANCD2, FANCG, FANCI, GEN1, RAD51, RAD54L, UBE2T, USP1, XRCC2, XRCC3 |
| Independent Repair Enzymes/Polymerases | DNA2, POLQ |
| Independent Repair Enzymes/Polymerases–Base excision repair | POLD1, POLD4, POLE2 |
| Independent Repair Enzymes/Polymerases—Cell Cycle and Signaling | MAD2L2 |
| Independent Repair Enzymes/Polymerases–Nucleotide Excision Repair | POLR2D, POLR2H |
| Independent Repair Enzymes/Polymerases–Translesion Synthesis | HLTF |
| Mismatch Repair | MSH2 |
| Mismatch Repair–Translesion Synthesis | RFC4 |
| Non-homologous End Joining—Cell Cycle and Signaling | PRKDC |
| Translesion Synthesis | RAD18 |
| Housekeeping | SF3FA3 |
| (B) Genes downregulated in tumors. | |
| Pathway | Genes |
| Apoptosis | AKT3, BCL2, NFKB2, PIK3R1 |
| Base Excision Repair | NEIL1, NEIL2, OGG1 |
| Cell Cycle and Signaling | ABL1, CCND2, CDKN1A, EGFR |
| Checkpoint Activation | TIPIN |
| Homologous Recombination and Fanconi Anemia | RAD51B, WRN |
| Independent Repair Enzymes/Polymerases | ALKBH3, CRY1, POLK, REV1 |
| Independent Repair Enzymes/Polymerases–Translesion Synthesis | POLI |
| Mismatch Repair | MSH3 |
| Non-homologous End Joining | LIG4 |
| Nucleotide Excision Repair | XPA |
| Nucleotide Excision Repair—Apoptosis | PTEN |
| Housekeeping | COG7, NUBP1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Custódio, N.; Savisaar, R.; Carvalho, C.; Bak-Gordon, P.; Ribeiro, M.I.; Tavares, J.; Nunes, P.B.; Peixoto, A.; Pinto, C.; Escudeiro, C.; et al. Expression Profiling in Ovarian Cancer Reveals Coordinated Regulation of BRCA1/2 and Homologous Recombination Genes. Biomedicines 2022, 10, 199. https://doi.org/10.3390/biomedicines10020199
Custódio N, Savisaar R, Carvalho C, Bak-Gordon P, Ribeiro MI, Tavares J, Nunes PB, Peixoto A, Pinto C, Escudeiro C, et al. Expression Profiling in Ovarian Cancer Reveals Coordinated Regulation of BRCA1/2 and Homologous Recombination Genes. Biomedicines. 2022; 10(2):199. https://doi.org/10.3390/biomedicines10020199
Chicago/Turabian StyleCustódio, Noélia, Rosina Savisaar, Célia Carvalho, Pedro Bak-Gordon, Maria I. Ribeiro, Joana Tavares, Paula B. Nunes, Ana Peixoto, Carla Pinto, Carla Escudeiro, and et al. 2022. "Expression Profiling in Ovarian Cancer Reveals Coordinated Regulation of BRCA1/2 and Homologous Recombination Genes" Biomedicines 10, no. 2: 199. https://doi.org/10.3390/biomedicines10020199
APA StyleCustódio, N., Savisaar, R., Carvalho, C., Bak-Gordon, P., Ribeiro, M. I., Tavares, J., Nunes, P. B., Peixoto, A., Pinto, C., Escudeiro, C., Teixeira, M. R., & Carmo-Fonseca, M. (2022). Expression Profiling in Ovarian Cancer Reveals Coordinated Regulation of BRCA1/2 and Homologous Recombination Genes. Biomedicines, 10(2), 199. https://doi.org/10.3390/biomedicines10020199