
Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study Design of Ang II-Dependent Hypertension
2.2. Peripheral Quantitative Computed Tomography (pQCT) Analysis
2.3. Bone Histology and Histomorphometry
2.4. Statistical Analysis
3. Results
3.1. Effects of Ang II and Losartan Administration on Systolic Blood Pressure, Body Weight, Glomerular Filtration Rate, Heart/Body Weight Ratio, Kidney/Body Weight Ratio and Serological Parameters
3.2. Effects of Ang II and Losartan Administration on 24 h Diuresis, Urinary Sodium, Calcium, and Phosphate Excretion
3.3. pQCT Analysis of Bone Parameters after Four Weeks of Treatment with Ang II and Losartan
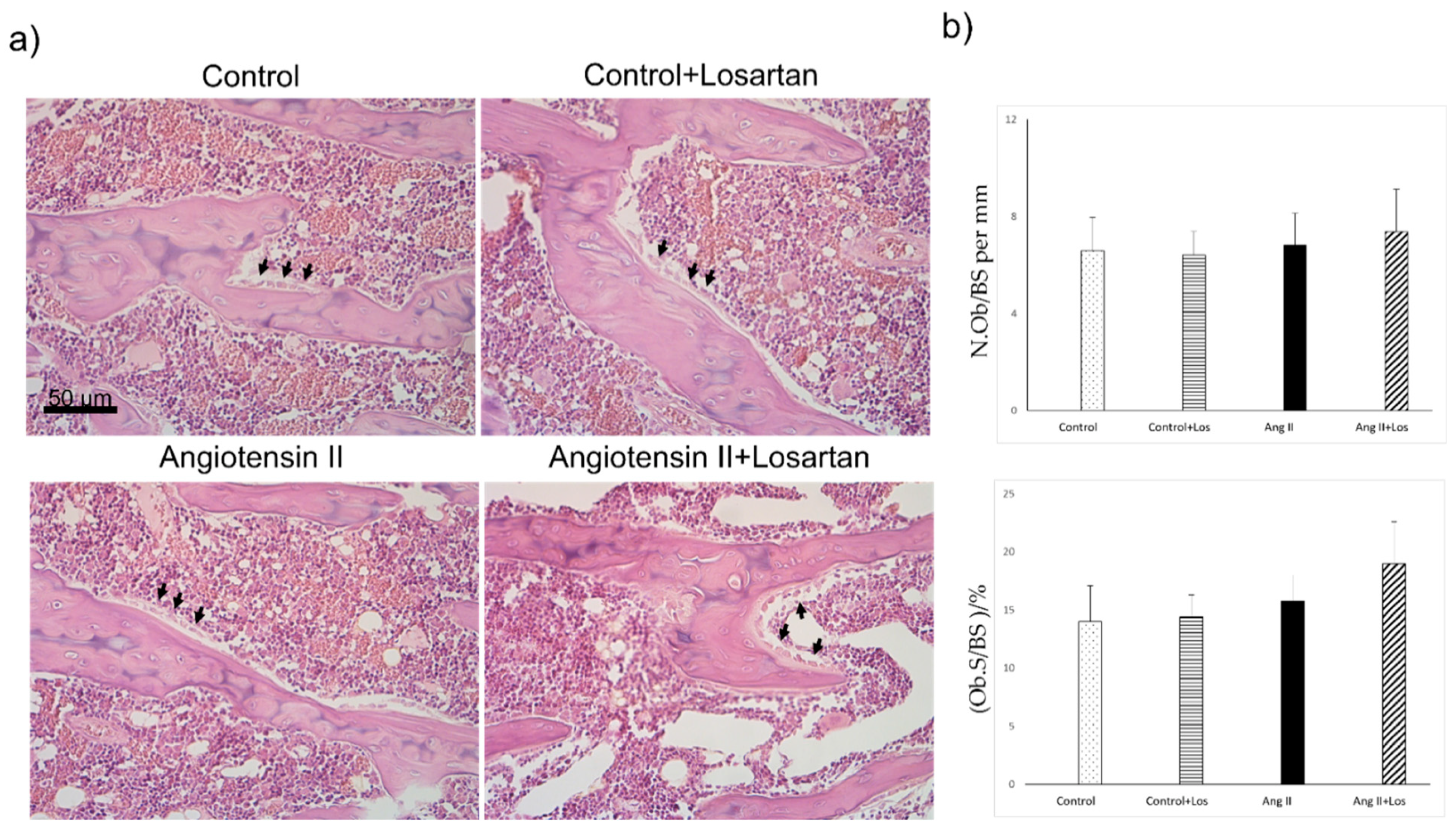
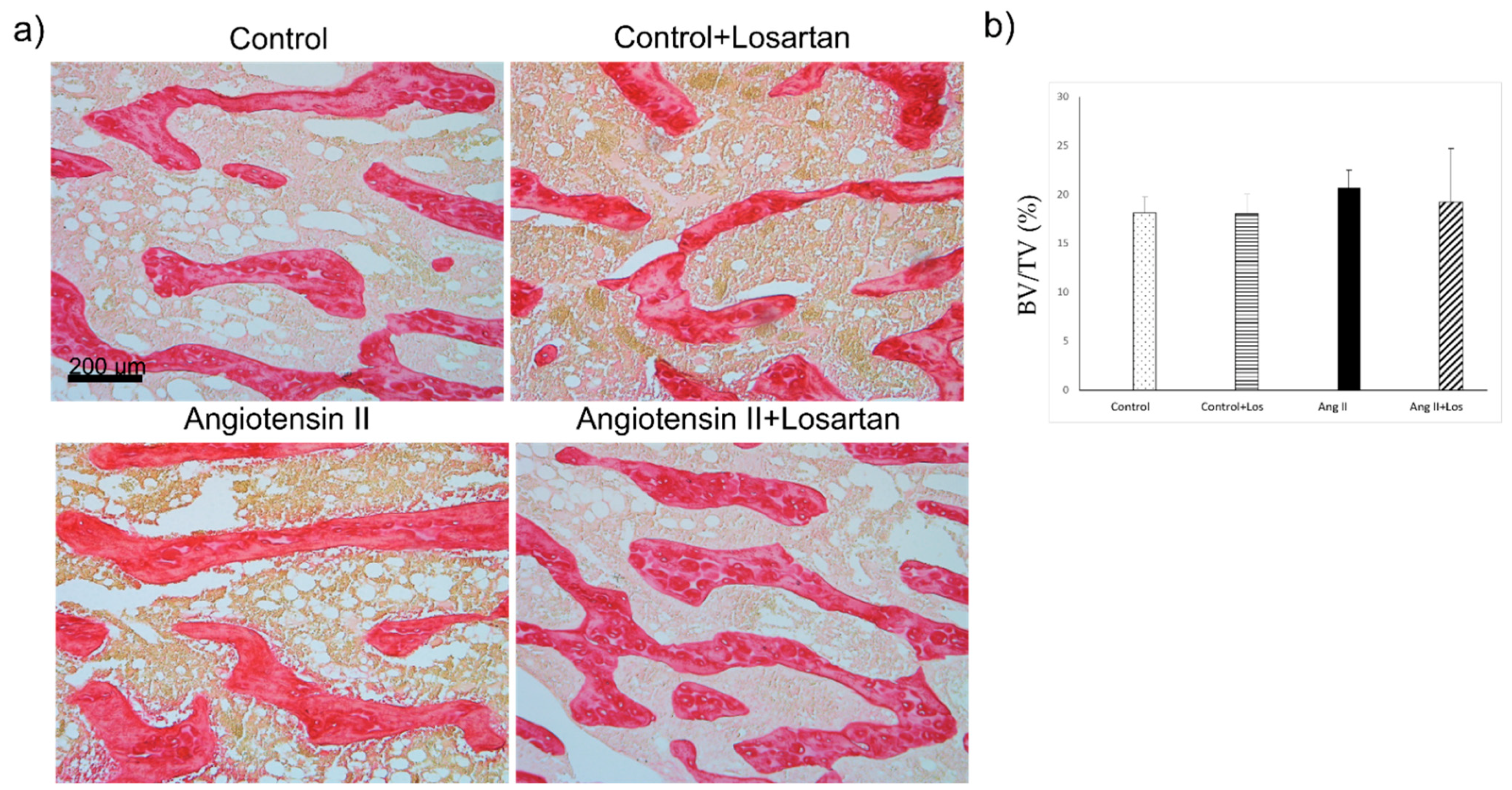
3.4. Histomorphometric Analysis of Bone Parameters after Four Weeks of Treatment with Ang II and Losartan
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Farhat, G.N.; Cauley, J.A. The link between osteoporosis and cardiovascular disease. Clin. Cases Miner. Bone Metab. 2008, 5, 19–34. [Google Scholar] [PubMed]
- Canoy, D.; Harvey, N.C.; Prieto-Alhambra, D.; Cooper, C.; Meyer, H.E.; Asvold, B.O.; Nazarzadeh, M.; Rahimi, K. Elevated blood pressure, antihypertensive medications and bone health in the population: Revisiting old hypotheses and exploring future research directions. Osteoporos. Int. 2022, 33, 315–326. [Google Scholar] [CrossRef] [PubMed]
- London, G.M. Soft bone–hard arteries: A link? Kidney Blood Press. Res. 2011, 34, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Asotra, K.; Fitzpatrick, L.A.; Qiao, J.H.; Wilkin, D.J.; Detrano, R.C.; Dunstan, C.R.; Shah, P.K.; Rajavashisth, T.B. Calcification in atherosclerosis: Bone biology and chronic inflammation at the arterial crossroads. Proc. Natl. Acad. Sci. USA 2003, 100, 11201–11206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.V.; Chapleau, M.W.; Harwani, S.C.; Abboud, F.M. The immune system and hypertension. Immunol. Res. 2014, 59, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Byon, C.H.; Chen, Y. Molecular mechanisms of vascular calcification in chronic kidney disease: The link between bone and the vasculature. Curr. Osteoporos. Rep. 2015, 13, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Grobbee, D.E.; Hackeng, W.H.L.; Birkenhager, J.C.; Hofman, A. Raised plasma intact parathyroid hormone concentrations in young people with mildly raised blood pressure. Br. Med. J. 1998, 296, 814–816. [Google Scholar] [CrossRef] [Green Version]
- Butt, D.A.; Alharty, R.; Leu, R.; Cheung, A.M. Hypertension, antihypertensive drugs and the risk of fractures. Clinic. Rev. Bone Mineral Metab. 2015, 13, 160–172. [Google Scholar] [CrossRef]
- Ilić, K.; Obradović, N.; Vujasinović-Stupar, N. The relationship among hypertension, antihypertensive medications, and osteoporosis: A narrative review. Calcif. Tissue Int. 2013, 92, 217–227. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.M.; Roks, A.J.M.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-Angiotensin-Aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, N.R.; Masi, S.; Taddei, S. The renin-angiotensin-aldosterone system: A crossroad from arterial hypertension to heart failure. Heart Fail. Rev. 2020, 25, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gómez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Z.; Luo, M.; Cheng, Z.; Wang, R.; Liu, Q.; Lv, D.; Yan, J.; Shang, F.; Luo, S.; et al. NLRP3 inflammasome contributes to endothelial dysfunction in angiotensin II-induced hypertension in mice. Microvasc. Res. 2022, 143, 104384. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Stella, A.; Zerbini, G.; Pelucchi, S.; Zatti, G.; di Gioia, C.R.T. Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. Int. J. Mol. Sci. 2021, 22, 13678. [Google Scholar] [CrossRef]
- Castoldi, G.; di Gioia, C.; Giollo, F.; Carletti, R.; Bombardi, C.; Antoniotti, M.; Roma, F.; Zerbini, G.; Stella, A. Different regulation of miR-29a-3p in glomeruli and tubules in an experimental model of angiotensin II-dependent hypertension: Potential role in renal fibrosis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 335–342. [Google Scholar] [CrossRef]
- Castoldi, G.; Di Gioia, C.R.T.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. MiR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J Cell Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.-X.; Han, Q.-Y.; Li, W.-J.; Zhang, Y.-L.; Du, J.; Xia, Y.-L.; Li, H.-H. Activation of the cardiac proteasome promotes angiotensin II-induced hypertrophy by down-regulation of ATRAP. J. Mol. Cell. Cardiol. 2015, 79, 303–314. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Zhou, Y.; Guan, X.; Chen, X.; Yu, M.; Wang, C.; Chen, X.; Shi, J.; Liu, T.; Wang, H. Angiotensin II/Angiotensin II receptor blockade affects osteoporosis via AT1/AT2-mediated cAMP-dependent PKA pathway. Cell Tissues Organs 2017, 204, 25–37. [Google Scholar] [CrossRef]
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. The ACE-2/Ang 1-7/Mas cascade enhances bone structure and metabolism following angiotensin-II type 1 receptor blockade. Europ. J. Pharmacol. 2017, 807, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Asaba, Y.; Ito, M.; Fumoto, T.; Watanabe, K.; Fukuhara, R.; Takeshita, S.; Nimura, Y.; Ishida, J.; Fukamizu, A.; Ikeda, K. Activation of renin-angiotensin system induces osteoporosis independently of hypertension. J. Bone Mineral. Res. 2009, 24, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nakagami, H.; Osako, M.K.; Hanayama, R.; Kunugiza, Y.; Kizawa, T.; Tomita, T.; Yoshikawa, H.; Ogihara, T.; Morishita, R. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008, 22, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.S.; Zhang, Y.; Li, X.L.; Wu, S.Y.; Diao, T.Y.; Hai, R.; Deng, H.W. Involvement of the skeletal renin-angiotensin system in age-related osteoporosis in ageing mice. Biosci. Biotechnol. Biochem. 2012, 76, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.S.; Zhang, Y.; Wu, S.Y.; Diao, T.Y.; Gebru, Y.A.; Deng, H.W. Early molecular response of bone to obstructive nephropathy induced by unilateral ureteral obstruction in mice. Nephrology 2012, 17, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Ito, M.; Fumoto, T.; Fukuhara, R.; Ishida, J.; Fukamizu, A.; Ikeda, K. Physiological function of the angiotensin AT1a receptor in bone remodeling. J. Bone Mineral Res. 2011, 26, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.-L.; Sha, N.-N.; Shu, B.; Zhao, Y.-J.; Wang, X.-L.; Xiao, H.-H.; Shi, Q.; Wong, M.-S.; Wang, Y.-J. Differential response of bone and kidney to ACEI in db/db mice: A potential effect of captopril on accelerating bone loss. Bone 2017, 97, 222–232. [Google Scholar] [CrossRef]
- Diao, T.Y.; Pan, H.; Gu, S.S.; Chen, X.; Zhang, F.Y.; Wong, M.S.; Zhang, Y. Effects of angiotensin-converting enzyme inhibitor, captopril, on bone of mice with streptozotocin-induced type 1 diabetes. J. Bone Miner. Metab. 2014, 32, 261–270. [Google Scholar] [CrossRef]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Colzani, M.; Barzaghi, F.; Stella, A.; Zerbini, G.; Perseghin, G.; Zatti, G.; di Gioia, C.R.T. Sodium-glucose cotransporter 2 inhibition prevents renal fibrosis in cyclosporine nephropathy. Acta Diabetol. 2021, 58, 1059–1070. [Google Scholar] [CrossRef]
- Castoldi, G.; di Gioia, C.R.T.; Roma, F.; Carletti, R.; Manzoni, G.; Stella, A.; Zerbini, G.; Perseghin, G. Activation of angiotensin type 2 (AT2) receptors prevents myocardial hypertrophy in Zucker diabetic fatty rats. Acta Diabetol. 2019, 56, 97–104. [Google Scholar] [CrossRef]
- Palmisano, B.; Labella, R.; Donsante, S.; Remoli, C.; Spica, E.; Coletta, I.; Farinacci, G.; Dello Spedale Venti, M.; Saggio, I.; Serafini, M.; et al. GsαR201C and estrogen reveal different subsets of bone marrow adiponectin expressing osteogenic cells. Bone Res. 2022, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, F.P.; Meilahn, E.; Zmuda, J.M.; Cauley, J.A. High blood pressure and bone-mineral loss in elderly white women: A prospective study. Study of Osteoporotic Fractures Research Group. Lancet 1999, 354, 971–975. [Google Scholar] [CrossRef]
- Cappuccio, F.P.; Kalaitzidis, R.; Duneclift, S.; Eastwood, J.B. Unravelling the links between calcium excretion, salt intake, hypertension, kidney stones and bone metabolism. J. Nephrol. 2000, 13, 169–177. [Google Scholar] [PubMed]
- Ye, Z.; Lu, H.; Liu, P. Association between essential hypertension and bone mineral density: A systematic review and meta-analysis. Oncotarget 2017, 8, 68916–68927. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dixit, M.P.; Chen, R.; Dixit, N.M.; Collins, J.F.; Ghishan, F.K. Effects of angiotensin II on NaPi-IIa-co-trasporter expression and activity in rat renal cortex. Biochim. Biophys. Acta 2004, 1667, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, E.; Hannemann, A.; Rettig, R.; Lieb, W.; Nauck, M.; Pallauf, A.; Bildingmaier, M.; Beuschlein, F.; Wallaschofski, H.; Reincke, M. A high aldosterone to renin ratio is associated with high serum parathyroid hormone concentrations in the general population. J. Clin. Endocrinol. Metab. 2014, 99, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Koiwa, F.; Komukai, D.; Hirose, M.; Yoshimura, A.; Ando, R.; Sakaguchi, T.; Komatsu, Y.; Shinoda, T.; Inaguma, D.; Joki, N.; et al. Influence of renin-angiotensin system on serum parathyroid hormone levels in uremic patients. Clin. Exp. Nephrol. 2012, 16, 130–135. [Google Scholar] [CrossRef]
- Catena, C.; Colussi, G.L.; Brosolo, G.; Bertin, N.; Novello, M.; Palomba, A.; Sechi, L.A. Salt, Aldosterone, and Parathyroid Hormone: What Is the Relevance for Organ Damage? Int. J. Endocrinol. 2017, 2017, 4397028. [Google Scholar] [CrossRef] [Green Version]
- Radloff, J.; Pagitz, M.; Andrukhova, O.; Oberbauer, R.; Burgener, I.A.; Erben, R.G. Aldosterone is positively associated with circulating FGF23 levels in chronic kidney disease across four species and may drive FGF23 secretion directly. Front. Physiol. 2021, 12, 649921. [Google Scholar] [CrossRef]
- Zhang, B.; Umbach, A.T.; Chen, H.; Yan, J.; Fakhri, H.; Fajol, A.; Salker, M.S.; Spichtig, D.; Daryadel, A.; Wagner, C.A.; et al. Up-regulation of FGF23 release by aldosterone. Biochem. Biophys. Res. Commun. 2016, 470, 384–390. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Kirchhoff, F.; Nespor, J.; Richter, B.; Soetje, B.; Klintschar, M.; Heineke, J.; Haffner, D. Fibroblast growth factor 23 is induced by an activated renin-angiotensin-aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol. Dial. Transplant. 2018, 33, 1722–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portales-Castillo, I.; Simic, P. PTH, FGF-23, Klotho and Vitamin D as regulators of calcium and phosphorus: Genetics, epigenetics and beyond. Front. Endocrinol. 2022, 13, 992666. [Google Scholar] [CrossRef] [PubMed]
- Ide, N.; Ye, R.; Courbebaisse, M.; Olauson, H.; Densmore, M.J.; Larsson, T.E.; Hanai, J.I.; Lanske, B. In vivo evidence for an interplay of FGF23/Klotho/PTH axis on the phosphate handling in renal proximal tubules. Am. J. Physiol. Renal Physiol. 2018, 315, F1261–F1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhatre, K.N.; Wakula, P.; Klein, O.; Bisping, E.; Volkl, J.; Pieske, B.; Heinzel, F.R. Crosstalk between FGF23- and angiotensin II-mediated Ca2+ signaling in pathological cardiac hypertrophy. Cell. Mol. Life Sci. 2018, 75, 4403–4416. [Google Scholar] [CrossRef]
- Akagi, T.; Mukai, T.; Mito, T.; Kawahara, K.; Tsuji, S.; Fujita, S.; Uchida, H.A.; Morita, Y. Effect of angiotensin II on bone erosion and systemic bone loss in mice with tumor necrosis factor-mediated arthritis. Int. J. Mol. Sci. 2020, 21, 4145. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | Control + Los | Ang II | Ang II + Los |
|---|---|---|---|---|
| SBP, mmHg | 140.5 ± 3.3 | 135.0 ± 5.0 | 202.1 ± 3.0 ‡§ζ | 142.5 ± 3.2 |
| BW, g | 459.1 ± 13.0 | 433.0 ± 5.2 | 409.7 ± 13.6 † | 423.8 ± 9.4 |
| GFR, ml/min | 4.28 ± 0.23 | 3.58 ± 0.07 | 3.35 ± 0.36 * | 4.15 ± 0.28 |
| Heart/Body Weight, mg/g | 2.81 ± 0.06 | 2.66 ± 0.13 | 3.93 ± 0.11 ‡§ζ | 2.71 ± 0.05 |
| Kidney/Body Weight, mg/g | 3.34 ± 0.05 | 3.51 ± 0.06 | 4.06 ± 0.09 ‡δζ | 3.42 ± 0.13 |
| Plasma | ||||
| Glucose (mg/dL) | 139± 4.08 | 142.1 ± 8.15 | 150.5 ± 5.03 | 137.2 ± 7.84 |
| Alkaline phosphatase (U/L) | 194.3 ± 13.7 | 194.4 ± 19.5 | 207.3 ± 6.97 | 226.7 ± 21.2 |
| Sodium (mEq/L) | 140.7 ± 0.38 | 141.3 ± 0.56 | 138.0 ± 0.93 †δ^ | 141.1 ± 1.03 |
| Potassium (mEq/L) | 3.95 ± 0.13 | 4.59 ± 0.33 | 3.16 ± 0.13 *δ& | 4.75 ± 0.54 * |
| Calcium (mg/dL) | 9.83 ± 0.10 | 9.96 ± 0.09 | 9.79 ± 0.17 | 10.0 ± 0.13 |
| Phosphate (mg/dL) | 5.72 ± 0.25 | 6.27 ± 0.46 | 4.84 ± 0.38 *°^ | 6.14 ± 0.45 |
| Uric acid (mg/dL) | 0.72 ± 0.15 | 0.72 ± 0.14 | 0.95 ± 0.18 | 0.94 ± 0.20 |
| Cholesterol (mg/dL) | 71.88 ± 3.96 | 70.2 ± 4.23 | 69.31 ± 3.51 | 71.14 ± 2.09 |
| Triglycerides (mg/dL) | 89.81 ± 4.89 | 98.6 ± 6.86 | 87.58 ± 21.32 | 116.34 ± 14.54 |
| Parameters | Control | Control + Los | Ang II | Ang II + Los |
|---|---|---|---|---|
| Tb.BMD (mg/cm3) | 257.0 ± 11.3 | 277.6 ± 19.7 | 251.9 ± 9.9 | 291.7 ± 10.2 |
| Tb.Area (mm2) | 20.47 ± 1.08 | 20.80 ± 1.53 | 21.66 ± 1.07 | 20.35 ± 0.71 |
| Ct.BMD (mg/cm3) | 1334.6 ± 4.8 | 1336.2 ± 8.5 | 1332.2 ± 6.6 | 1331.8 ± 5.3 |
| Ct.Area (mm2) | 4.75 ± 0.16 | 4.67 ± 0.10 | 4.52 ± 0.18 | 4.71 ± 0.16 |
| Tot.Area (mm2) | 6.91 ± 0.24 | 6.75 ± 0.19 | 6.54 ± 0.25 | 6.90 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castoldi, G.; Carletti, R.; Ippolito, S.; Villa, I.; Palmisano, B.; Bolamperti, S.; Rubinacci, A.; Zerbini, G.; Meani, M.; Zatti, G.; et al. Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone. Biomedicines 2022, 10, 2928. https://doi.org/10.3390/biomedicines10112928
Castoldi G, Carletti R, Ippolito S, Villa I, Palmisano B, Bolamperti S, Rubinacci A, Zerbini G, Meani M, Zatti G, et al. Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone. Biomedicines. 2022; 10(11):2928. https://doi.org/10.3390/biomedicines10112928
Chicago/Turabian StyleCastoldi, Giovanna, Raffaella Carletti, Silvia Ippolito, Isabella Villa, Biagio Palmisano, Simona Bolamperti, Alessandro Rubinacci, Gianpaolo Zerbini, Michela Meani, Giovanni Zatti, and et al. 2022. "Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone" Biomedicines 10, no. 11: 2928. https://doi.org/10.3390/biomedicines10112928
APA StyleCastoldi, G., Carletti, R., Ippolito, S., Villa, I., Palmisano, B., Bolamperti, S., Rubinacci, A., Zerbini, G., Meani, M., Zatti, G., & di Gioia, C. R. T. (2022). Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone. Biomedicines, 10(11), 2928. https://doi.org/10.3390/biomedicines10112928