
Role of Translationally Controlled Tumor Protein (TCTP) in the Development of Hypertension and Related Diseases in Mouse Models
Abstract
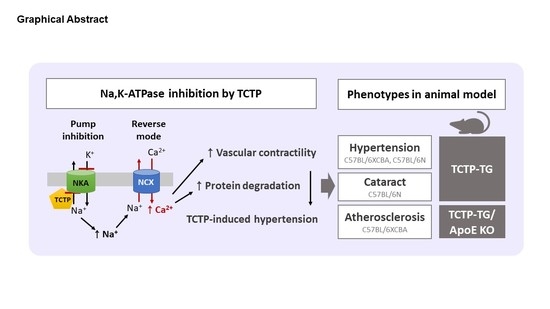
1. Introduction
2. Pathophysiological Consequences of Na,K-ATPase Inhibition
2.1. Inhibition of Pump Activity of Na,K-ATPase by CTS
2.2. Signal Transduction of Na,K-ATPase by CTS
2.3. Ouabain-Induced Hypertension and Related Diseases
3. TCTP, as an Intracellular Na,K-ATPase Suppressor
4. Pathophysiology of Hypertension in TCTP-TG
4.1. Systemic Arterial Hypertension by Na,K-ATPase Inhibition in TCTP-TG
4.2. Upregulation of RhoA/Rho Kinase Signaling in TCTP-TG
4.3. Elevation of the Expression of Peroxiredoxin 3 (Prx3) and Heat Shock Protein 25 (Hsp25) in TCTP-TG
5. Pathophysiology of Cataractogenesis in TCTP-TG
6. Pathophysiology of Atherosclerosis in TCTP-TG and TCTP+/− Mice
6.1. Exacerbation of Atherosclerosis by TCTP-Induced Hypertension in TCTP-TG/ApoE KO
6.2. Aggravation of Atherosclerosis by TCTP-Induced Survival of Macrophages in TCTP+/− Mice
7. Role of TCTP in Hypertension-Related Diseases
7.1. Obesity
7.2. Heart Failure
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al Ghorani, H.; Götzinger, F.; Böhm, M.; Mahfoud, F. Arterial Hypertension—Clinical Trials Update 2021. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Carrillo-Larco, R.M.; Danaei, G.; Riley, L.M.; Paciorek, C.J.; Stevens, G.A.; Gregg, E.W.; Bennett, J.E.; Solomon, B.; Singleton, R.K.; et al. NCD Risk Factor Collaboration (NCD-RisC) Worldwide Trends in Hypertension Prevalence and Progress in Treatment and Control from 1990 to 2019: A Pooled Analysis of 1201 Population-Representative Studies with 104 Million Participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Kućmierz, J.; Frąk, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Molecular Interactions of Arterial Hypertension in Its Target Organs. Int. J. Mol. Sci. 2021, 22, 9669. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Chen, L.; Hamlyn, J.M.; Leenen, F.H.H.; Lingrel, J.B.; Wier, W.G.; Zhang, J. Pivotal Role of A2 Na+ Pumps and Their High Affinity Ouabain Binding Site in Cardiovascular Health and Diseaysse. J. Physiol. 2016, 594, 6079–6103. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.M.; Manunta, P.; Hamlyn, J.M.; Chen, S.; Bohen, E.; Yeun, J.; Haddy, F.J.; Pamnani, M.B. Long-Term Ouabain Administration Produces Hypertension in Rats. Hypertension 1993, 22, 178–187. [Google Scholar] [CrossRef]
- Manunta, P.; Rogowski, A.C.; Hamilton, B.P.; Hamlyn, J.M. Ouabain-Induced Hypertension in the Rat: Relationships among Plasma and Tissue Ouabain and Blood Pressure. J. Hypertens. 1994, 12, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Buckalew, V.M. Endogenous Digitalis-like Factors: An Overview of the History. Front. Endocrinol. 2015, 6, 49. [Google Scholar] [CrossRef]
- Lingrel, J.B. The Physiological Significance of the Cardiotonic Steroid/Ouabain-Binding Site of the Na,K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, J.I. The Physiological and Clinical Importance of Sodium Potassium ATPase in Cardiovascular Diseases. Curr. Opin. Pharmacol. 2016, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Scheiner-Bobis, G. The Na+, K+-ATPase: More than Just a Sodium Pump. Cardiovasc. Res. 2011, 89, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990. [Google Scholar] [CrossRef]
- Xie, Z.; Xie, J. The Na/K-ATPase-Mediated Signal Transduction as a Target for New Drug Development. Front. Biosci. 2005, 10, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, P.F.; Orellana, A.M.M.; Nakao, V.W.; de Souza Port’s, N.M.; Quintas, L.E.M.; Kawamoto, E.M.; Scavone, C. The Janus Face of Ouabain in Na+/K+-ATPase and Calcium Signalling in Neurons. Br. J. Pharmacol. 2022, 179, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.B.; Delpire, E. Sodium Transporters in Human Health and Disease. Front. Physiol. 2020, 11, 588664. [Google Scholar] [CrossRef] [PubMed]
- Biondo, E.D.; Spontarelli, K.; Ababioh, G.; Méndez, L.; Artigas, P. Diseases Caused by Mutations in the Na+/K+ Pump A1 Gene ATP1A1. Am. J. Physiol. Cell Physiol. 2021, 321, C394–C408. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, K.; Braendgaard, H.; Sidenius, P.; Larsen, J.S.; Nørgaard, A. Diabetes Decreases Na+-K+ Pump Concentration in Skeletal Muscles, Heart Ventricular Muscle, and Peripheral Nerves of Rat. Diabetes 1987, 36, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and Exogenous Cardiac Glycosides: Their Roles in Hypertension, Salt Metabolism, and Cell Growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.F.; Hollenberg, N.K.; Graves, S.W. Sodium Pump Inhibition and Regional Expression of Sodium Pump Alpha-Isoforms in Lens. Hypertension 1999, 34, 1168–1174. [Google Scholar] [CrossRef]
- Blaustein, M.P. Endogenous Ouabain: Role in the Pathogenesis of Hypertension. Kidney Int. 1996, 49, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Delamere, N.A.; Tamiya, S. Expression, Regulation and Function of Na,K-ATPase in the Lens. Prog. Retin. Eye Res. 2004, 23, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Askari, A. The Other Functions of the Sodium Pump. Cell Calcium 2019, 84, 102105. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and Epidermal Growth Factor Receptor in the Signal-Transducing Function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-Mediated Inter-Receptor Cross-Talk between the Na+/K+-ATPase and the Epidermal Growth Factor Receptor Relays the Signal from Ouabain to Mitogen-Activated Protein Kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, G.; Zhao, A.; Bondeva, T.; Hirszel, P.; Balla, T. Inhibition of Na,K-ATPase Activates PI3 Kinase and Inhibits Apoptosis in LLC-PK1 Cells. Biochem. Biophys. Res. Commun. 2001, 285, 46–51. [Google Scholar] [CrossRef]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase Tethers Phospholipase C and IP3 Receptor into a Calcium-Regulatory Complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and Exogenous Cardiac Glycosides and Their Mechanisms of Action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef]
- Manunta, P.; Hamilton, J.; Rogowski, A.C.; Hamilton, B.P.; Hamlyn, J.M. Chronic Hypertension Induced by Ouabain but Not Digoxin in the Rat: Antihypertensive Effect of Digoxin and Digitoxin. Hypertens. Res. 2000, 23, S77–S85. [Google Scholar] [CrossRef]
- Goto, A.; Yamada, K.; Yagi, N.; Yoshioka, M.; Sugimoto, T. Physiology and Pharmacology of Endogenous Digitalis-like Factors. Pharmacol. Rev. 1992, 44, 377–399. [Google Scholar]
- Bommer, U.-A.; Thiele, B.-J. The Translationally Controlled Tumour Protein (TCTP). Int. J. Biochem. Cell Biol. 2004, 36, 379–385. [Google Scholar] [CrossRef]
- Pinkaew, D.; Fujise, K. Fortilin: A Potential Target for the Prevention and Treatment of Human Diseases. Adv. Clin. Chem. 2017, 82, 265–300. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.-A.; Kawakami, T. Role of TCTP in Cell Biological and Disease Processes. Cells 2021, 10, 2290. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, M.; Kim, M.-J.; Kim, J.; Moon, J.; Lim, J.-S.; Kim, M.; Lee, K. Translationally Controlled Tumor Protein Interacts with the Third Cytoplasmic Domain of Na,K-ATPase Alpha Subunit and Inhibits the Pump Activity in HeLa Cells. J. Biol. Chem. 2004, 279, 49868–49875. [Google Scholar] [CrossRef]
- Assrir, N.; Malard, F.; Lescop, E. Structural Insights into TCTP and Its Interactions with Ligands and Proteins. Results Probl. Cell Differ. 2017, 64, 9–46. [Google Scholar] [CrossRef]
- Bommer, U.-A. The Translational Controlled Tumour Protein TCTP: Biological Functions and Regulation. Results Probl. Cell Differ. 2017, 64, 69–126. [Google Scholar] [CrossRef]
- Lee, H.-J.; Song, K.-H.; Oh, S.J.; Kim, S.; Cho, E.; Kim, J.; Park, Y.G.; Lee, K.-M.; Yee, C.; Song, S.-H.; et al. Targeting TCTP Sensitizes Tumor to T Cell-Mediated Therapy by Reversing Immune-Refractory Phenotypes. Nat. Commun. 2022, 13, 2127. [Google Scholar] [CrossRef]
- Bommer, U.-A.; Telerman, A. Dysregulation of TCTP in Biological Processes and Diseases. Cells 2020, 9, 1632. [Google Scholar] [CrossRef]
- Koide, Y.; Kiyota, T.; Tonganunt, M.; Pinkaew, D.; Liu, Z.; Kato, Y.; Hutadilok-Towatana, N.; Phongdara, A.; Fujise, K. Embryonic Lethality of Fortilin-Null Mutant Mice by BMP-Pathway Overactivation. Biochim. Biophys. Acta 2009, 1790, 326–338. [Google Scholar] [CrossRef]
- Kim, M.-J.; Kwon, J.-S.; Suh, S.H.; Suh, J.-K.; Jung, J.; Lee, S.-N.; Kim, Y.-H.; Cho, M.-C.; Oh, G.T.; Lee, K. Transgenic Overexpression of Translationally Controlled Tumor Protein Induces Systemic Hypertension via Repression of Na+,K+-ATPase. J. Mol. Cell. Cardiol. 2008, 44, 151–159. [Google Scholar] [CrossRef]
- Maeng, J.; Sheverdin, V.; Shin, H.; Ha, I.; Bae, S.S.; Yang-Yen, H.-F.; Lee, K. Up-Regulation of Rhoa/Rho Kinase Pathway by Translationally Controlled Tumor Protein in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2014, 15, 10365–10376. [Google Scholar] [CrossRef]
- Kim, M.-J.; Lyu, J.; Sohn, K.-B.; Kim, M.; Cho, M.-C.; Joo, C.-K.; Lee, K. Over-Expression of Translationally Controlled Tumor Protein in Lens Epithelial Cells Seems to Be Associated with Cataract Development. Transgenic Res. 2009, 18, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Shin, H.K.; Yang-Yen, H.-F.; Lee, M.S.; Lee, C.H.; Park, S.-J.; Kim, K.-J.; Lee, K.; Kim, S.H. Osteoclastogenic Activity of Translationally-Controlled Tumor Protein (TCTP) with Reciprocal Repression of P21. FEBS Lett. 2014, 588, 4026–4031. [Google Scholar] [CrossRef]
- Kim, M.; Choe, Y.; Lee, H.; Jeon, M.-G.; Park, J.-H.; Noh, H.S.; Cheon, Y.-H.; Park, H.J.; Park, J.; Shin, S.J.; et al. Blockade of Translationally Controlled Tumor Protein Attenuated the Aggressiveness of Fibroblast-like Synoviocytes and Ameliorated Collagen-Induced Arthritis. Exp. Mol. Med. 2021, 53, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Maeng, J.; Ryu, J.; Shin, H.; Kim, M.; Oh, G.T.; Lee, M.-Y.; Lee, K. Hypertension Resulting from Overexpression of Translationally Controlled Tumor Protein Increases the Severity of Atherosclerosis in Apolipoprotein E Knock-out Mice. Transgenic Res. 2012, 21, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Pinkaew, D.; Le, R.J.; Chen, Y.; Eltorky, M.; Teng, B.-B.; Fujise, K. Fortilin Reduces Apoptosis in Macrophages and Promotes Atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1519–H1529. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-C.; Xie, L.; Langdon, J.M.; Myers, A.C.; Oh, S.-Y.; Zhu, Z.; Macdonald, S.M. The Effects of Overexpression of Histamine Releasing Factor (HRF) in a Transgenic Mouse Model. PLoS ONE 2010, 5, e11077. [Google Scholar] [CrossRef]
- Jeon, Y.; Choi, J.-Y.; Jang, E.-H.; Seong, J.K.; Lee, K. Overexpression of Translationally Controlled Tumor Protein Ameliorates Metabolic Imbalance and Increases Energy Expenditure in Mice. Int. J. Obes. 2021, 45, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xiong, Z.; Zhang, S.; Song, J.; Huang, Y.; Thornton, A.M.; Wang, H.; Yang, X.-F. CD25high T Cells with a Prolonged Survival Inhibit Development of Diabetes. Int. J. Immunopathol. Pharmacol. 2008, 21, 767–780. [Google Scholar] [CrossRef]
- Cai, W.; Fujita, T.; Hidaka, Y.; Jin, H.; Suita, K.; Shigeta, M.; Kiyonari, H.; Umemura, M.; Yokoyama, U.; Sadoshima, J.; et al. Translationally Controlled Tumor Protein (TCTP) Plays a Pivotal Role in Cardiomyocyte Survival through a Bnip3-Dependent Mechanism. Cell Death Dis. 2019, 10, 549. [Google Scholar] [CrossRef]
- Chunhacha, P.; Pinkaew, D.; Sinthujaroen, P.; Bowles, D.E.; Fujise, K. Fortilin Inhibits P53, Halts Cardiomyocyte Apoptosis, and Protects the Heart against Heart Failure. Cell Death Discov. 2021, 7, 310. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Pinkaew, D.; Doan, H.Q.; Jacob, R.B.; Verma, S.K.; Friedman, H.; Peterson, A.C.; Kuyumcu-Martinez, M.N.; McDougal, O.M.; Fujise, K. Fortilin Potentiates the Peroxidase Activity of Peroxiredoxin-1 and Protects against Alcohol-Induced Liver Damage in Mice. Sci. Rep. 2016, 6, 18701. [Google Scholar] [CrossRef] [PubMed]
- Pinkaew, D.; Chattopadhyay, A.; King, M.D.; Chunhacha, P.; Liu, Z.; Stevenson, H.L.; Chen, Y.; Sinthujaroen, P.; McDougal, O.M.; Fujise, K. Fortilin Binds IRE1α and Prevents ER Stress from Signaling Apoptotic Cell Death. Nat. Commun. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Leong, X.-F.; Ng, C.-Y.; Jaarin, K. Animal Models in Cardiovascular Research: Hypertension and Atherosclerosis. BioMed Res. Int. 2015, 2015, 528757. [Google Scholar] [CrossRef] [PubMed]
- Cargnelli, G.; Rossi, G.P.; Pessina, A.C.; Luciani, S.; Debetto, P.; Ganten, D.; Peters, J.; Bova, S. Changes of Blood Pressure and Aortic Strip Contractile Responses to ET-1 of Heterozygous Female Transgenic Rats, TGR(MRen2)27. Pharmacol. Res. 1998, 37, 207–211. [Google Scholar] [CrossRef]
- McVeigh, G.E.; Plumb, R.; Hughes, S. Vascular Abnormalities in Hypertension: Cause, Effect, or Therapeutic Target? Curr. Hypertens. Rep. 2004, 6, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Different Effects of in Vivo Ouabain and Digoxin on Renal Artery Function and Blood Pressure in the Rat. Hypertens. Res. 2000, 23, S67–S76. [Google Scholar] [CrossRef]
- Wirth, A. Rho Kinase and Hypertension. Biochim. Biophys. Acta 2010, 1802, 1276–1284. [Google Scholar] [CrossRef]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and Inhibition of Myosin Phosphatase as Important Components in Hypertension in Vascular Smooth Muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium Sensitization of Smooth Muscle Mediated by a Rho-Associated Protein Kinase in Hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular-Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.S.; Suh, J.-K.; Kim, M.-J.; Kim, S.H.; Lee, K. Identification of Differentially Expressed Proteins in the Heart of Translationally Controlled Tumor Protein Over-Expressing Transgenic Mice. Biomed. Chromatogr. 2008, 22, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E. Contribution of Mitochondrial Oxidative Stress to Hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Ungvari, Z. Role of Mitochondrial Oxidative Stress in Hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Jeong, S.-J.; Park, J.-G.; Oh, G.T. Peroxiredoxins as Potential Targets for Cardiovascular Disease. Antioxidants 2021, 10, 1244. [Google Scholar] [CrossRef]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of Mitochondrial Peroxiredoxin-3 Prevents Left Ventricular Remodeling and Failure after Myocardial Infarction in Mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef]
- Ishizaka, N.; Aizawa, T.; Ohno, M.; Usui Si, S.; Mori, I.; Tang, S.-S.; Ingelfinger, J.R.; Kimura, S.; Nagai, R. Regulation and Localization of HSP70 and HSP25 in the Kidney of Rats Undergoing Long-Term Administration of Angiotensin II. Hypertension 2002, 39, 122–128. [Google Scholar] [CrossRef]
- Szmyd, L.; Schwartz, B. Association of Systemic Hypertension and Diabetes Mellitus with Cataract Extraction. A Case-Control Study. Ophthalmology 1989, 96, 1248–1252. [Google Scholar] [CrossRef]
- Yu, X.; Lyu, D.; Dong, X.; He, J.; Yao, K. Hypertension and Risk of Cataract: A Meta-Analysis. PLoS ONE 2014, 9, e114012. [Google Scholar] [CrossRef]
- Unakar, N.J.; Johnson, M. Lenticular Alterations in Hypertensive Rats. Exp. Eye Res. 1994, 59, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sargent, C.; Cangiano, J.L.; Berríos Cabán, G.; Marrero, E.; Martínez-Maldonado, M. Cataracts and Hypertension in Salt-Sensitive Rats. A Possible Ion Transport Defect. Hypertension 1987, 9, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sargent, C.; Estapé, E.S.; Fernández, N.; Irizarry, J.E.; Cangiano, J.L.; Candia, O.A. Altered Lens Short-Circuit Current in Adult Cataract-Prone Dahl Hypertensive Rats. Hypertension 1996, 28, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Delamere, N.A.; Tamiya, S. Lens Ion Transport: From Basic Concepts to Regulation of Na,K-ATPase Activity. Exp. Eye Res. 2009, 88, 140–143. [Google Scholar] [CrossRef]
- Lichtstein, D.; Gati, I.; Samuelov, S.; Berson, D.; Rozenman, Y.; Landau, L.; Deutsch, J. Identification of Digitalis-like Compounds in Human Cataractous Lenses. Eur. J. Biochem. 1993, 216, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.D.; Johar, K.; Vasavada, A. Causative and Preventive Action of Calcium in Cataracto-Genesis. Acta Pharmacol. Sin. 2004, 25, 1250–1256. [Google Scholar]
- Karlsson, J.O.; Andersson, M.; Kling-Petersen, A.; Sjöstrand, J. Proteolysis in Human Lens Epithelium Determined by a Cell-Permeable Substrate. Invest. Ophthalmol. Vis. Sci. 1999, 40, 261–264. [Google Scholar]
- Gupta, J.D.; Harley, J.D. Decreased Adenosine Triphosphatase Activity in Human Senile Cataractous Lenses. Exp. Eye Res. 1975, 20, 207–209. [Google Scholar] [CrossRef]
- Ziegler, T.; Abdel Rahman, F.; Jurisch, V.; Kupatt, C. Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells 2019, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Hollander, W. Role of Hypertension in Atherosclerosis and Cardiovascular Disease. Am. J. Cardiol. 1976, 38, 786–800. [Google Scholar] [CrossRef]
- Ketonen, J.; Merasto, S.; Paakkari, I.; Mervaala, E.M.A. High Sodium Intake Increases Vascular Superoxide Formation and Promotes Atherosclerosis in Apolipoprotein E-Deficient Mice. Blood Press. 2005, 14, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.; Taylor, W.R. Deoxycorticosterone Acetate Salt Hypertension in Apolipoprotein E−/− Mice Results in Accelerated Atherosclerosis: The Role of Angiotensin II. Hypertension 2008, 51, 218–224. [Google Scholar] [CrossRef]
- da Cunha, V.; Tham, D.M.; Martin-McNulty, B.; Deng, G.; Ho, J.J.; Wilson, D.W.; Rutledge, J.C.; Vergona, R.; Sullivan, M.E.; Wang, Y.-X.J. Enalapril Attenuates Angiotensin II-Induced Atherosclerosis and Vascular Inflammation. Atherosclerosis 2005, 178, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Standridge, J.B. Hypertension and Atherosclerosis: Clinical Implications from the ALLHAT Trial. Curr. Atheroscler. Rep. 2005, 7, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous Hypercholesterolemia and Arterial Lesions in Mice Lacking Apolipoprotein, E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-Deficient Mice Develop Lesions of All Phases of Atherosclerosis throughout the Arterial Tree. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Chen, J.-L. Inflammation May Be a Bridge Connecting Hypertension and Atherosclerosis. Med. Hypotheses 2005, 64, 925–929. [Google Scholar] [CrossRef]
- Barton, M.; Yanagisawa, M. Endothelin: 30 Years From Discovery to Therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef]
- Ivey, M.E.; Osman, N.; Little, P.J. Endothelin-1 Signalling in Vascular Smooth Muscle: Pathways Controlling Cellular Functions Associated with Atherosclerosis. Atherosclerosis 2008, 199, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-J.; Juan, C.-C.; Kwok, C.-F.; Hsu, Y.-P.; Shih, K.-C.; Chen, C.-C.; Ho, L.-T. Endothelin-1 Exacerbates Development of Hypertension and Atherosclerosis in Modest Insulin Resistant Syndrome. Biochem. Biophys. Res. Commun. 2015, 460, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, R.R.; Prasanna, G.; Dauphin, R.; Hulet, C.; Agarwal, N.; Yorio, T. Regulation of Na,K-ATPase Expression by Endothelin-1 in Transformed Human Ciliary Non-Pigmented Epithelial (HNPE) Cells. J. Ocul. Pharmacol. Ther. 2003, 19, 465–481. [Google Scholar] [CrossRef]
- Mandal, A.; Shahidullah, M.; Beimgraben, C.; Delamere, N.A. The Effect of Endothelin-1 on Src-Family Tyrosine Kinases and Na,K-ATPase Activity in Porcine Lens Epithelium. J. Cell. Physiol. 2011, 226, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, G.; Dibas, A.; Hulet, C.; Yorio, T. Inhibition of Na(+)/K(+)-Atpase by Endothelin-1 in Human Nonpigmented Ciliary Epithelial Cells. J. Pharmacol. Exp. Ther. 2001, 296, 966–971. [Google Scholar] [PubMed]
- Scheiner-Bobis, G.; Eva, A.; Kirch, U. Signalling Pathways Involving Sodium Pump Stimulate Endothelin-1 Secretion and Nitric Oxide Production in Endothelial Cells. Cell. Mol. Biol. 2006, 52, 58–63. [Google Scholar]
- Eva, A.; Kirch, U.; Scheiner-Bobis, G. Signaling Pathways Involving the Sodium Pump Stimulate NO Production in Endothelial Cells. Biochim. Biophys. Acta 2006, 1758, 1809–1814. [Google Scholar] [CrossRef]
- Predic, J.; Soskic, V.; Bradley, D.; Godovac-Zimmermann, J. Monitoring of Gene Expression by Functional Proteomics: Response of Human Lung Fibroblast Cells to Stimulation by Endothelin-1. Biochemistry 2002, 41, 1070–1078. [Google Scholar] [CrossRef]
- Saitoh, T.; Kishida, H.; Tsukada, Y.; Fukuma, Y.; Sano, J.; Yasutake, M.; Fukuma, N.; Kusama, Y.; Hayakawa, H. Clinical Significance of Increased Plasma Concentration of Macrophage Colony-Stimulating Factor in Patients with Angina Pectoris. J. Am. Coll. Cardiol. 2000, 35, 655–665. [Google Scholar] [CrossRef]
- Aoyama, M.; Kishimoto, Y.; Saita, E.; Ohmori, R.; Tanimoto, K.; Nakamura, M.; Kondo, K.; Momiyama, Y. High Plasma Levels of Fortilin in Patients with Coronary Artery Disease. Int. J. Mol. Sci. 2022, 23, 8923. [Google Scholar] [CrossRef]
- Piché, M.-E.; Tchernof, A.; Després, J.-P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a Molecular Understanding of Adaptive Thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Berbée, J.F.P.; Boon, M.R.; Khedoe, P.P.S.J.; Bartelt, A.; Schlein, C.; Worthmann, A.; Kooijman, S.; Hoeke, G.; Mol, I.M.; John, C.; et al. Brown Fat Activation Reduces Hypercholesterolaemia and Protects from Atherosclerosis Development. Nat. Commun. 2015, 6, 6356. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Vasileiou, P.V.; Papaspyropoulos, A.; Hazapis, O.; Petty, R.; Demaria, M.; Gorgoulis, V.G. Cellular Senescence and Cardiovascular Diseases: Moving to the “Heart” of the Problem. Physiol. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.-G.; Tang, Q.-Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Animal Models | Related Disease | Phenotypes | Ref. |
|---|---|---|---|
| Pathophysiological roles of TCTP | |||
| TCTP-TG | Hypertension | Development of systemic arterial hypertension and increased vascular contractility ↑ RhoA expression and phospho-myosin light chain (p-MLC) in aorta | [39,40] |
| Cataract | ↑ Incidence of cataract formation in lens ↑ Abnormal eye development | [41] | |
| Osteoporosis | ↓ Bone mass in femurs ↑ Osteoporotic features in femur bones, and osteoclast cell count | [42] | |
| Rheumatoid arthritis | ↑ Inflammatory responses, bone erosion, and cartilage destruction upon collagen-induced arthritis (CIA) | [43] | |
| TCTP+/− | Hypertension | ↓ RhoA expression and p-MLC in aorta | [40] |
| Osteoporosis | ↑ Bone mass in femurs ↓ Osteoporotic features in femur bones, and osteoclast cell count | [42] | |
| Rheumatoid arthritis | ↓ Synovial inflammation, bone erosion, cartilage damage, and osteoclastic bone resorption upon CIA | [43] | |
| TCTP-TG/ ApoE-KO | Atherosclerosis | ↑ Exacerbation of atherosclerotic lesion formation by high-fat diet without alteration in plasma lipid profiles, compared with ApoE-KO mice | [44] |
| TCTP+/− Ldlr−/−Apobec1−/− | Atherosclerosis | Similar lipid profiles and BP compared with TCTP+/+ Ldlr−/− Apobec1−/− mice ↓ Atherosclerotic lesion in aorta and macrophage numbers in lesions, compared to TCTP+/+ Ldlr−/− Apobec1−/− mice ↑ Bax expression and apoptosis of peritoneal macrophages in the intima layer, compared with TCTP+/+ Ldlr−/− Apobec1−/− mice | [45] |
| Clara cell-specific TCTP-TG | Allergic asthma | ↑ Allergic and asthmatic inflammation with increase in serum and bronchoalveolar lavage (BAL) IgE, interleukin-4 (IL-4), and eosinophil count upon ovalbumin (OVA) challenge ↑ TCTP secretion and macrophage counts in BAL fluids | [46] |
| Protective roles of TCTP | |||
| TCTP-TG | Obesity | ↑ Metabolic homeostasis under both normal and high-fat diet conditions with enhanced glucose tolerance and insulin sensitivity ↑ Energy expenditure with upregulation of uncoupling protein 1 (UCP1) in the brown adipose tissue (BAT) ↑ Adaptive thermogenesis of BAT after cold exposure | [47] |
| Tregs-specific TCTP-TG | Diabetes | ↑ Forkhead box protein P3(FOXP3) expression and prolonged survival of regulatory T cells (Tregs) ↓ Development of autoimmune diabetes by inhibiting the apoptosis of Tregs | [48] |
| Cardiomyocyte-specific TCTP-TG | Heart failure | ↓ Doxorubicin-induced cardiac dysfunction and Bcl-2 interacting protein 3 (Bnip3) induction ↓ Dihydroartemisinin-induced heart failure and cardiomyocyte death | [49] |
| Heart-specific TCTP-KO | Heart failure | Mice lacking TCTP in the heart die by 9 weeks of age because of severe heart failure and extensive cardiomyocyte apoptosis | [50] |
| Liver-specific TCTP-TG | Liver damage | ↑ Peroxiredoxin-1 (PRX1) activity in the liver and protection against alcohol, and ROS-mediated liver damage | [51] |
| Liver-specific TCTP-KO | Liver damage | ↓ Endoplasmic reticulum (ER) stress-induced liver failure and death by blocking apoptosis in the liver | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeng, J.; Lee, K. Role of Translationally Controlled Tumor Protein (TCTP) in the Development of Hypertension and Related Diseases in Mouse Models. Biomedicines 2022, 10, 2722. https://doi.org/10.3390/biomedicines10112722
Maeng J, Lee K. Role of Translationally Controlled Tumor Protein (TCTP) in the Development of Hypertension and Related Diseases in Mouse Models. Biomedicines. 2022; 10(11):2722. https://doi.org/10.3390/biomedicines10112722
Chicago/Turabian StyleMaeng, Jeehye, and Kyunglim Lee. 2022. "Role of Translationally Controlled Tumor Protein (TCTP) in the Development of Hypertension and Related Diseases in Mouse Models" Biomedicines 10, no. 11: 2722. https://doi.org/10.3390/biomedicines10112722
APA StyleMaeng, J., & Lee, K. (2022). Role of Translationally Controlled Tumor Protein (TCTP) in the Development of Hypertension and Related Diseases in Mouse Models. Biomedicines, 10(11), 2722. https://doi.org/10.3390/biomedicines10112722