
GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration


 , , ,
, , ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Transfection Consumables and Equipment
2.2. Rabbit Proof-of-Principle (PoP) Study
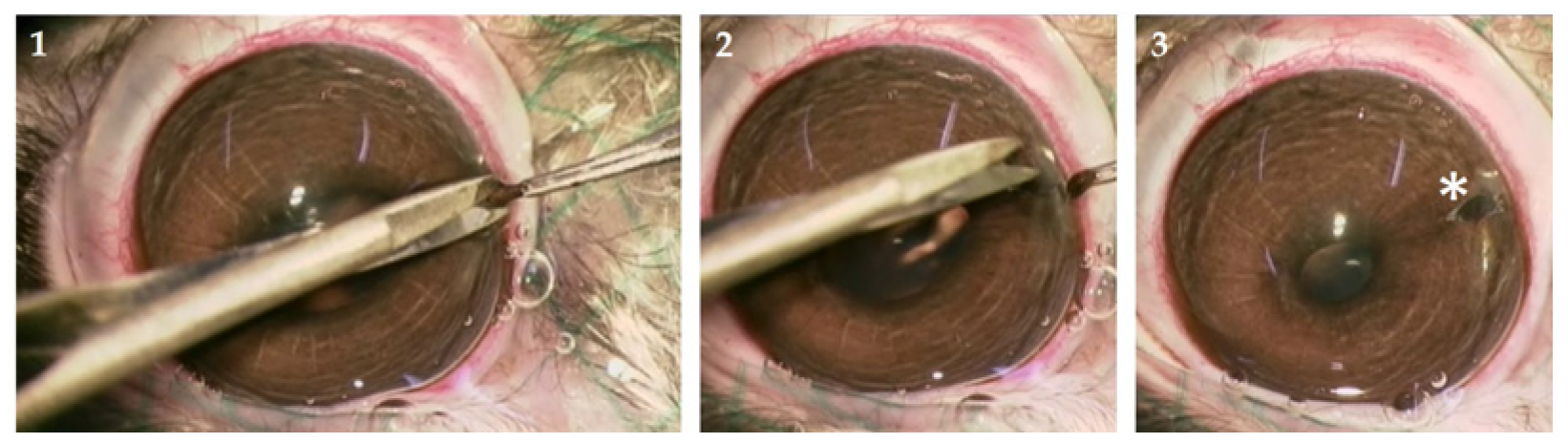
2.2.1. Animals and Iridectomy
2.2.2. Rabbit tIPE Production
2.2.3. Transplantation of tIPE in Rabbits
2.2.4. Intraocular Pressure (IOP) and Fundoscopy
2.2.5. Examination and Dissection of Rabbit Organs
2.3. Validation Run
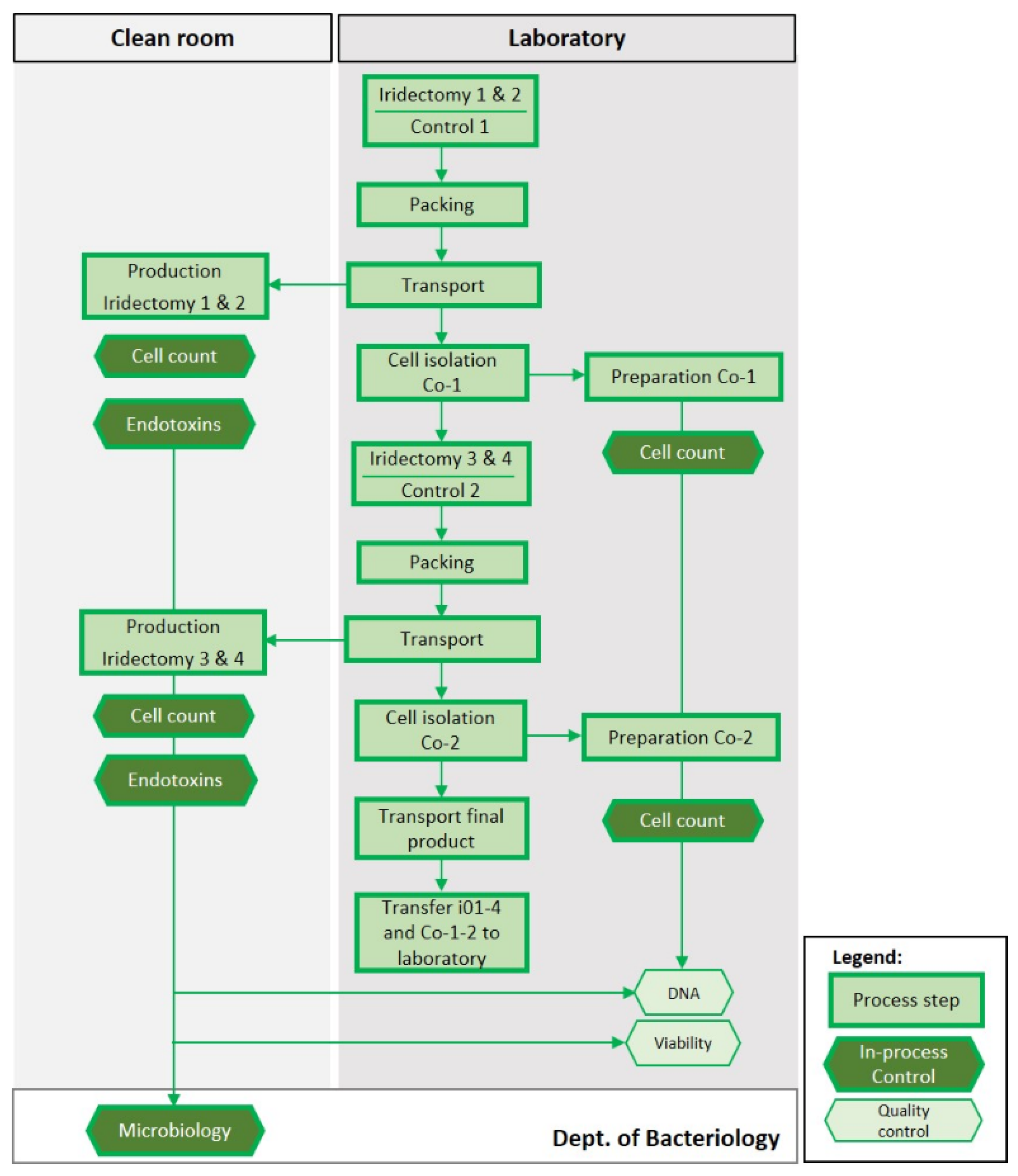
2.3.1. Human tIPE Production
2.3.2. Bioconfined Syringe Container
2.3.3. Cell Viability
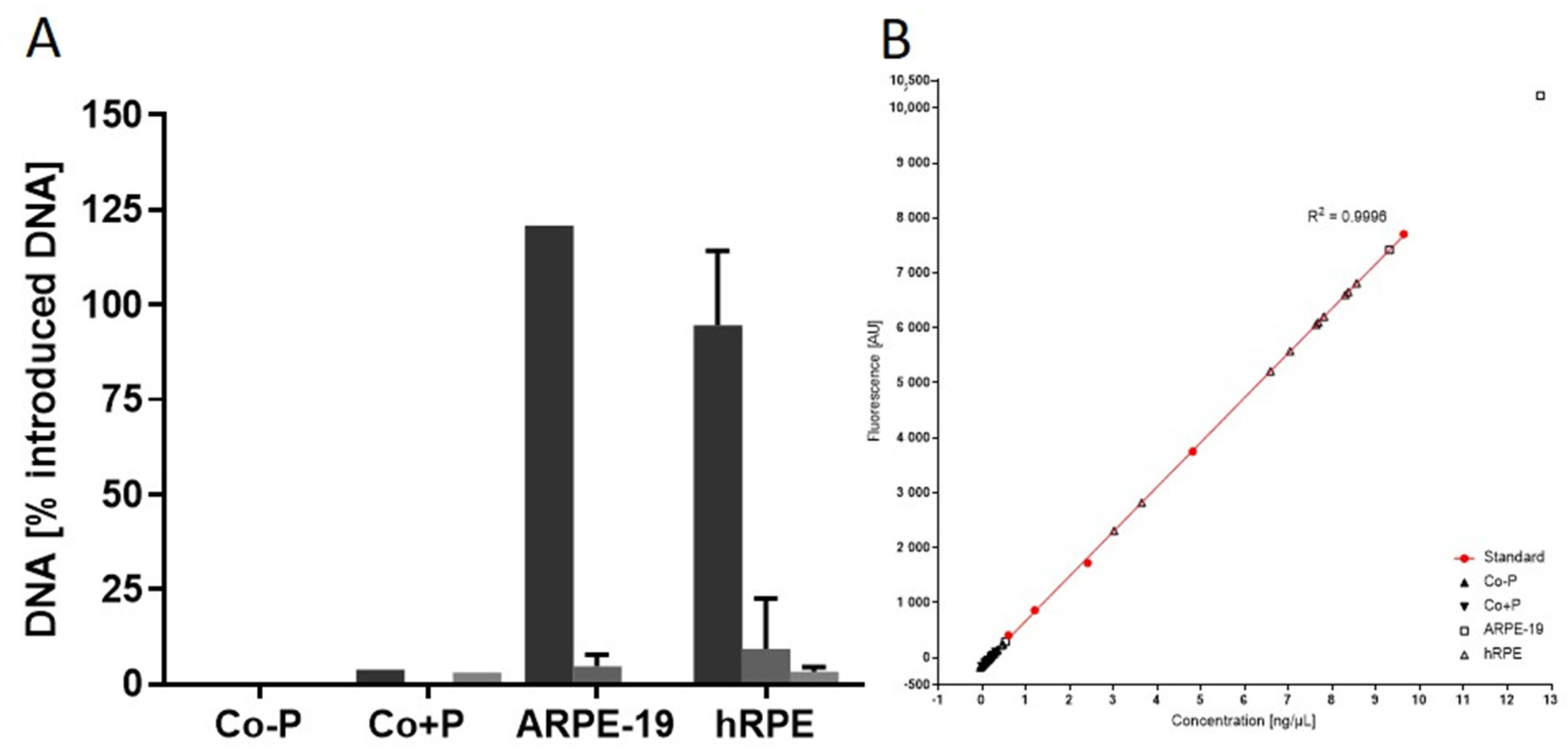
2.3.4. Contaminating Remnant Extracellular DNA
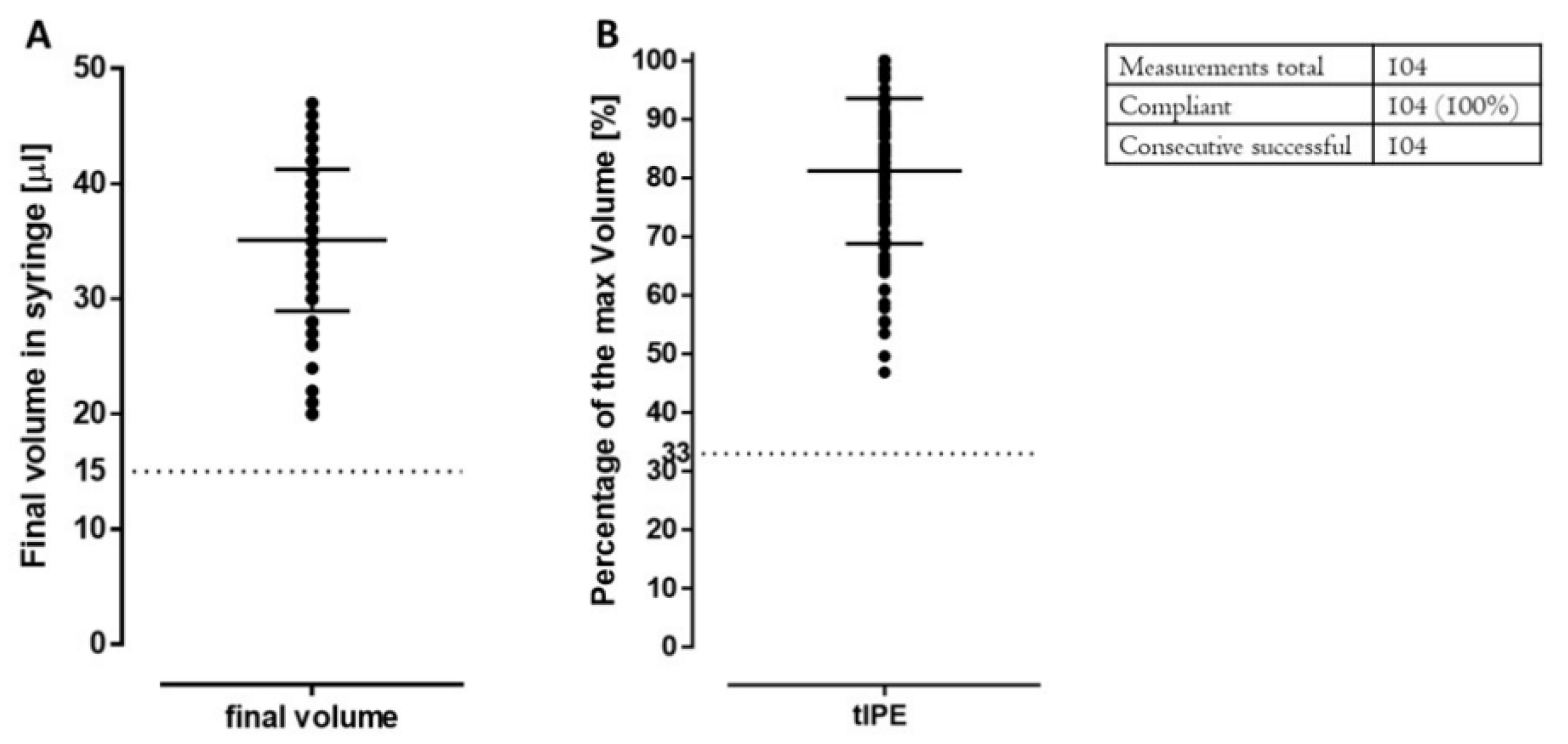
2.3.5. Product Volume and Appearance
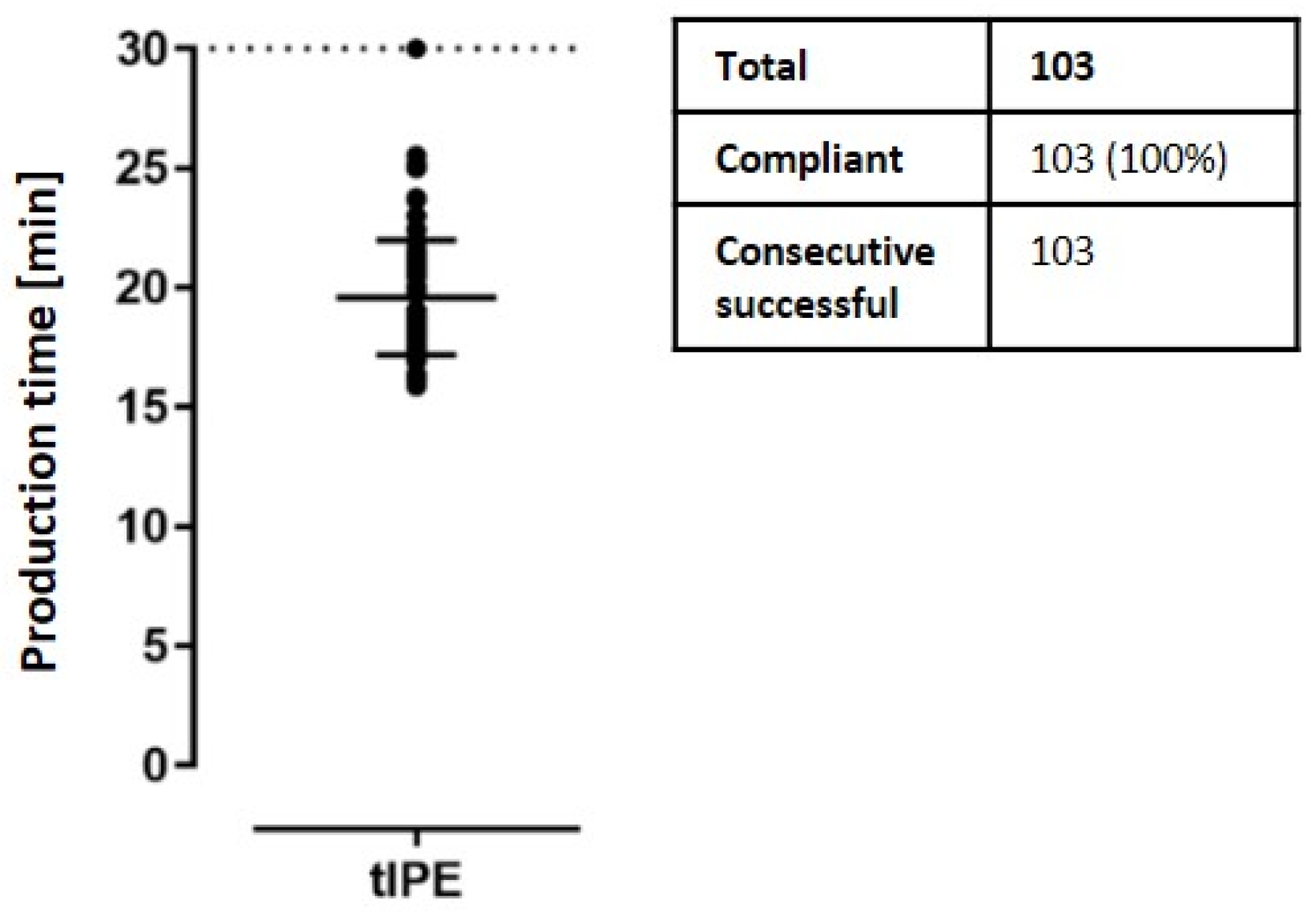
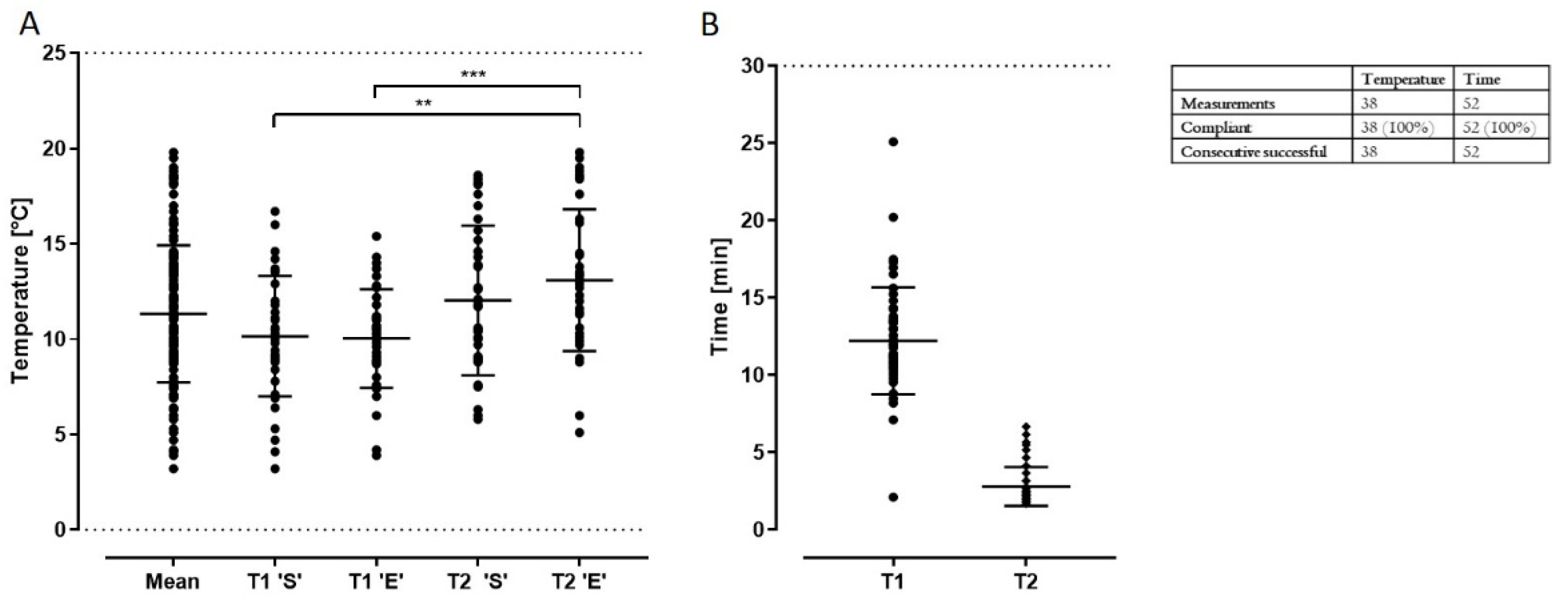
2.3.6. Production, Transport Time and Temperature
2.3.7. Endotoxin
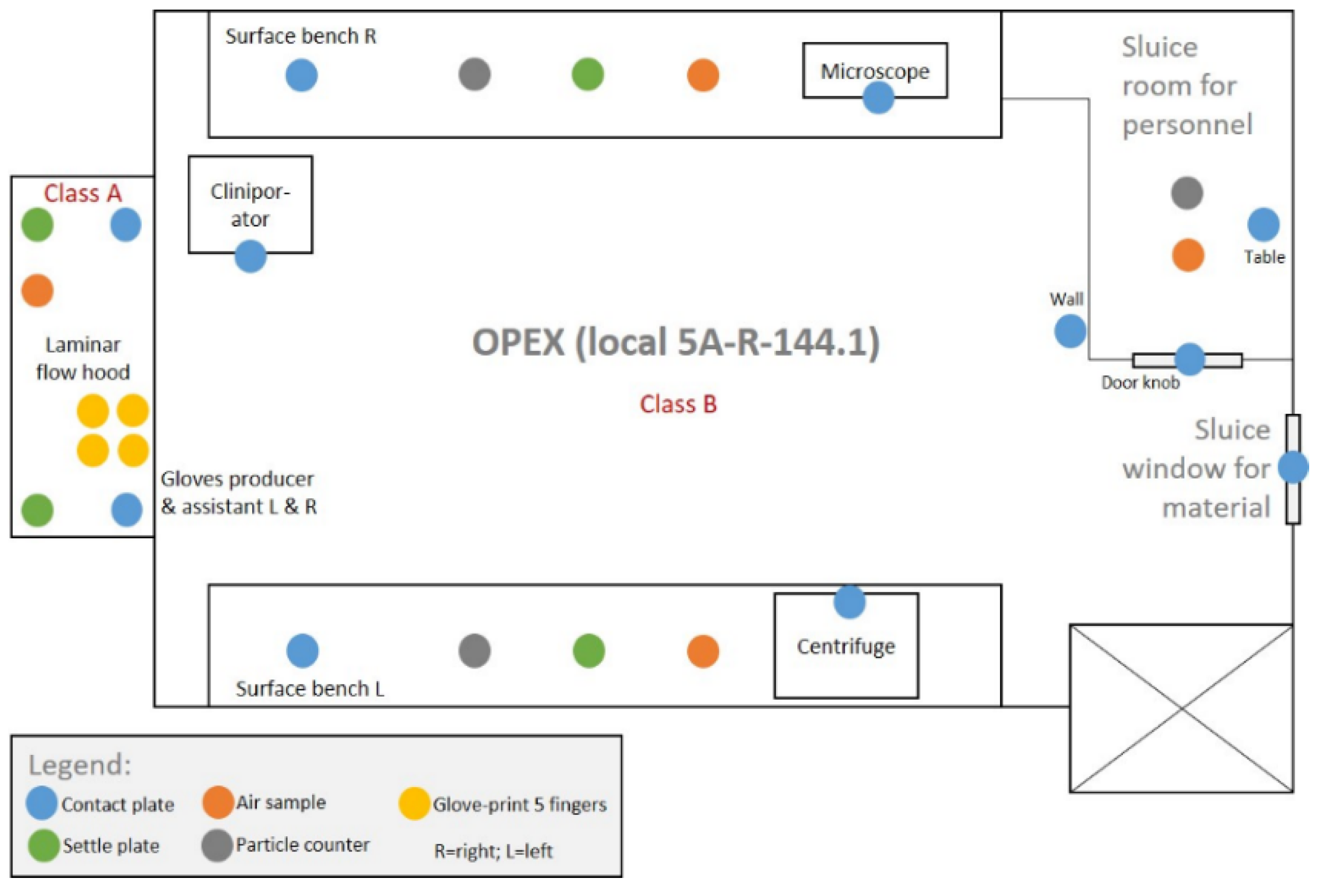
2.3.8. Microbiological and Environmental Controls
2.4. Statistics
3. Results
3.1. PoP Study—Validation Run in Rabbits
3.1.1. Surgical Complications and Side Effects
3.1.2. Fundus and IOP
3.1.3. Organ Phenotype
3.2. Bioconfined Syringe Container
3.3. GMP-Grade Validation Run
3.3.1. Framework—Purpose, Donors and Process
- Validate the process;
- Validate the cleaning procedure;
- Optimize the process under GMP-compliant conditions;
- Define release criteria;
- Collect sufficient in vitro data from human donor eyes to demonstrate quality of tIPE;
- Qualify the CliniporatorTM (performance qualification, PQ);
- Train (carrier, assistant, operator) and qualify personnel (operator).
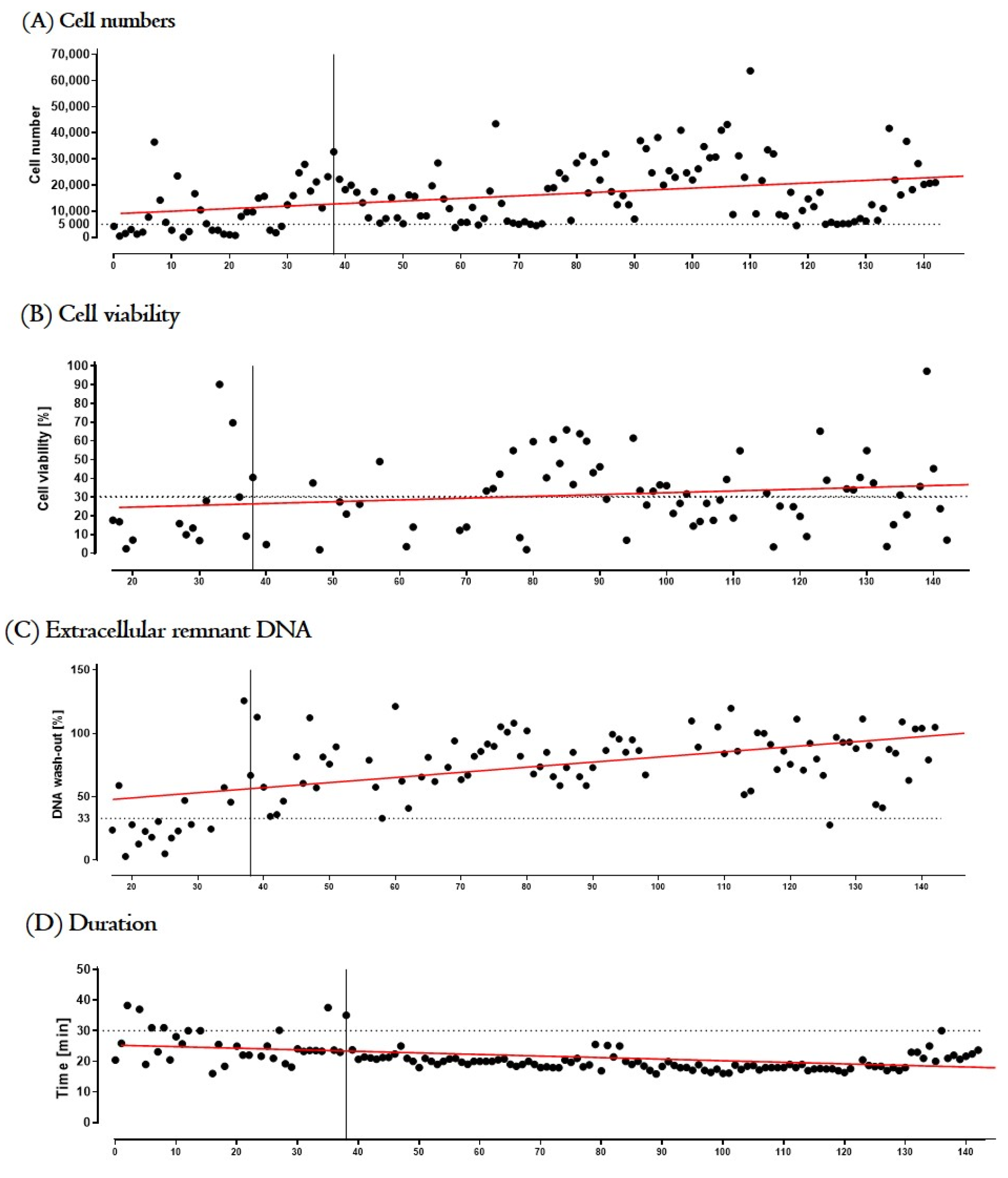
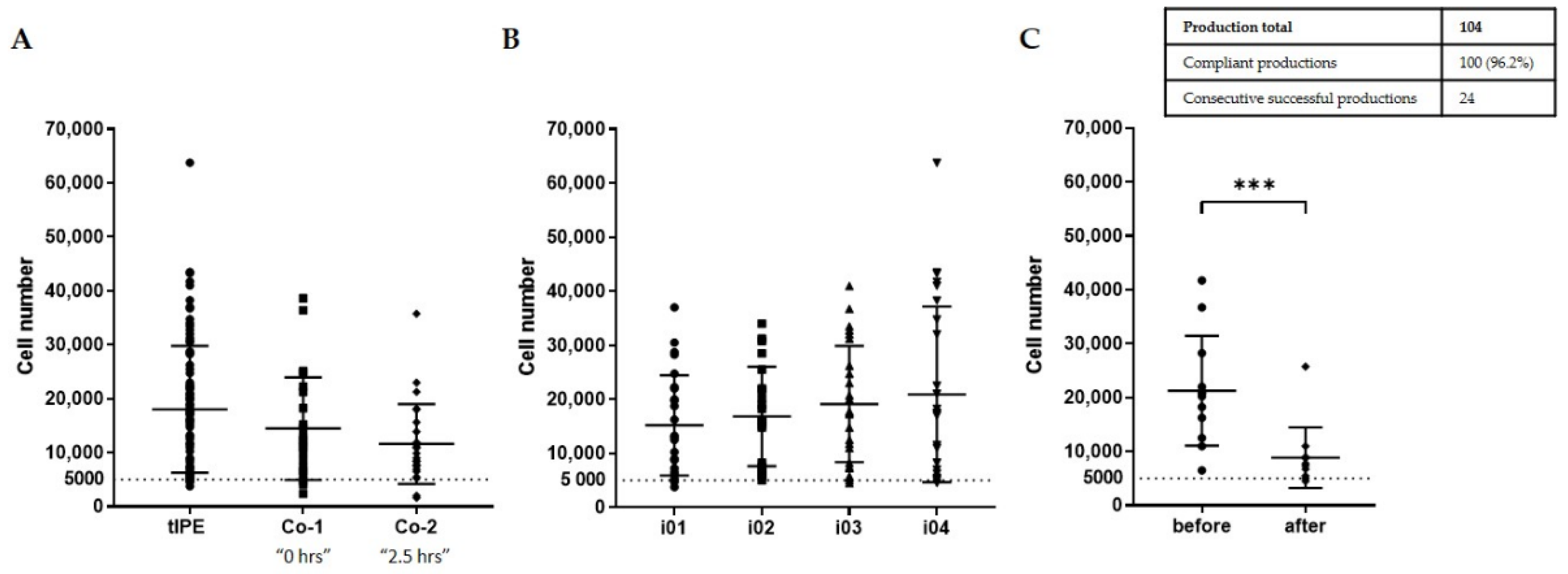
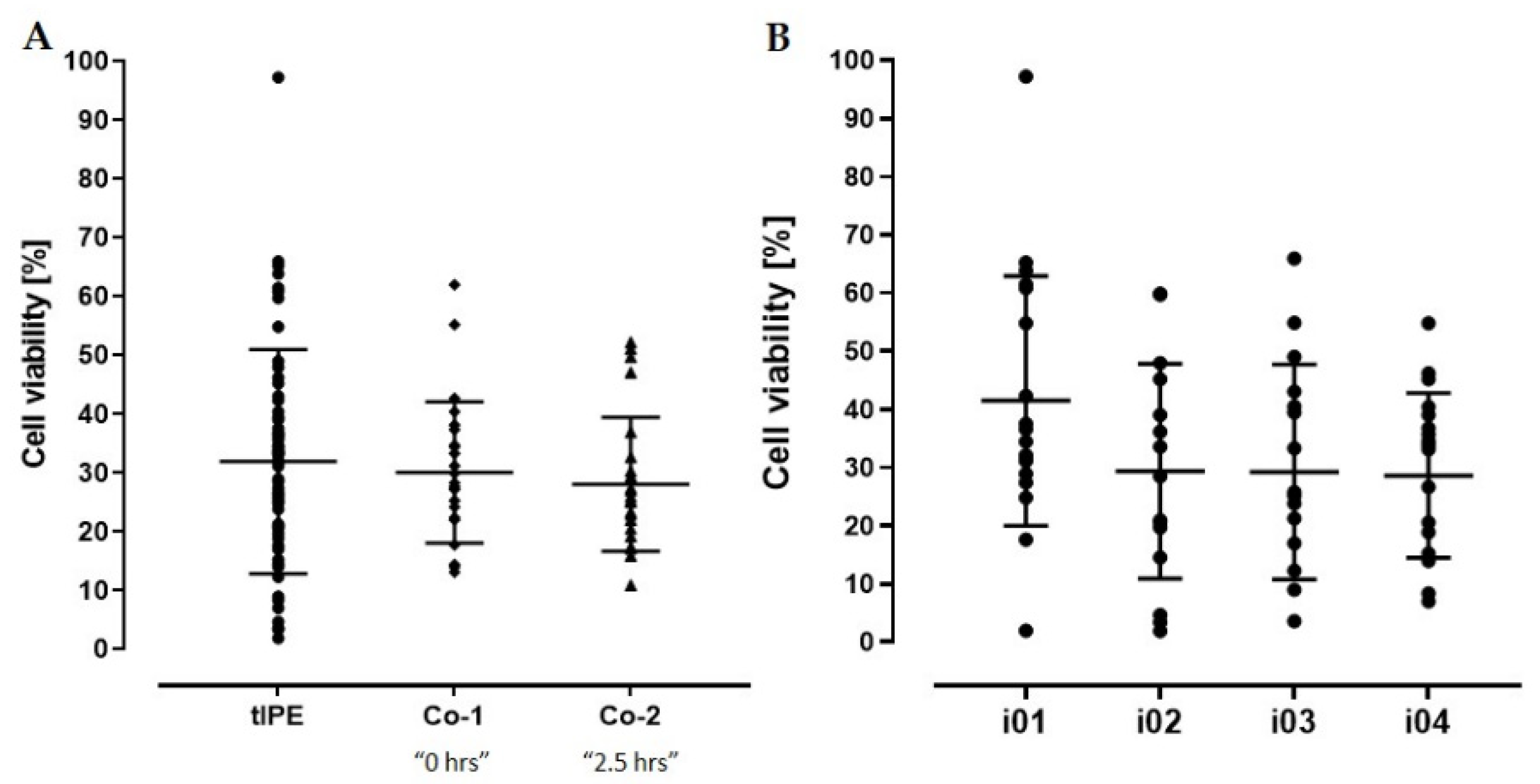
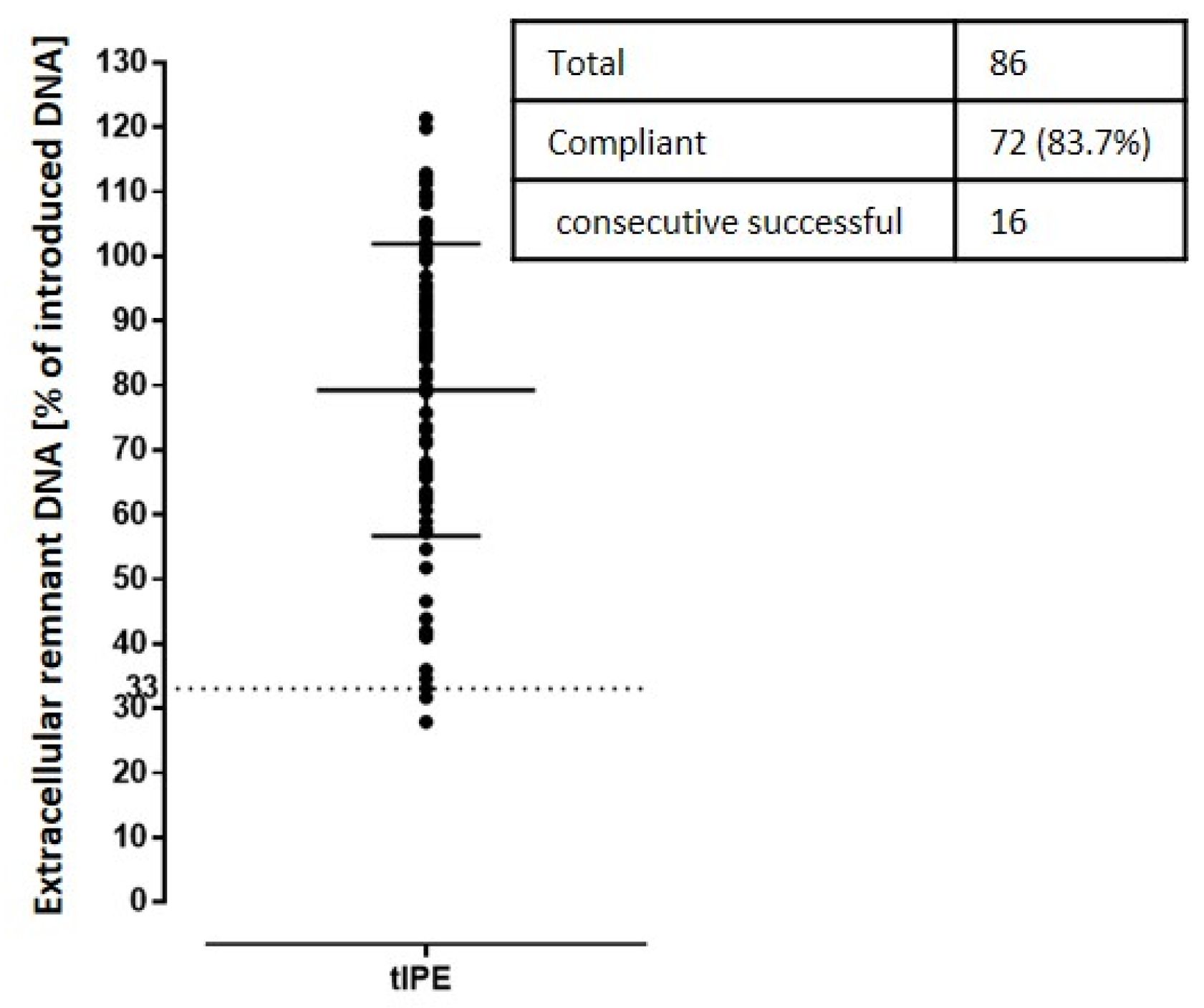
3.3.2. Process Optimization and Definition of Release Criteria
3.3.3. Validation Run
- Environmental sterility. Even though four biopsies were taken and four tIPE manufactured from each donor’s eye, during the ValRun only one environmental control per eye was analyzed for environmental microbiological contamination. In the case of a positive result, all four tIPEs from the respective eye were considered non-compliant. Results showed that 20 of 26 analyses (76.9%) were sterile with the last 8 consecutive analyses conforming. Particle analysis performed by the Labguard® system (Biomérieux, Petit Lancy, Switzerland) verified a GMP-conforming environment for 100 out of 104 productions (96.2%).
- Biopsy sterility. Analysis of the stroma from the iris biopsies showed that 96.2% (100) were sterile including the last 8 consecutive analyses.
- tIPE sterility. Analysis of the supernatant from the centrifugation after electroporation showed that 96.2% (100) of tIPE manufactured, including the last 8 consecutives, were sterile.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanna, E.; Rémuzat, C.; Auquier, P.; Toumi, M. Advanced therapy medicinal products: Current and future perspectives. J. Mark. Access Health Policy 2016, 4, 31036. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Viviani, M.; Fussenegger, M. Engineering precision therapies: Lessons and motivations from the clinic. Synth. Biol. 2021, 6, ysaa024. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S. Gene and Cell Therapy: Success Stories and Future Challenges. Mol. Ther. 2019, 27, 891–892. [Google Scholar] [CrossRef]
- Directive 2001/83/Ec of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use; European Union: Brussels, Belgium, 2012.
- Office, P. Regulation (Ec) No 1394/2007 of the European Parliament and of The Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) No 726/2004; European Union: Brussels, Belgium, 2007. [Google Scholar]
- López-Paniagua, M.; La Mata, A.; Galindo, S.; Blázquez, F.; Calonge, M.; Nieto-Miguel, T. Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics 2021, 13, 347. [Google Scholar] [CrossRef]
- Weitzel, J.; Pappa, H.; Banik, G.M.; Barker, A.R.; Bladen, E.; Chirmule, N.; DeFeo, J.; Devine, J.; Emrick, S.; Hout, T.K.; et al. Understanding Quality Paradigm Shifts in the Evolving Pharmaceutical Landscape: Perspectives from the USP Quality Advisory Group. AAPS J. 2021, 23, 112. [Google Scholar] [CrossRef]
- Jere, D.; Sediq, A.S.; Huwyler, J.; Vollrath, I.; Kardorff, M.; Mahler, H.-C. Challenges for Cell-Based Medicinal Products From a Pharmaceutical Product Perspective. J. Pharm. Sci. 2021, 110, 1900–1908. [Google Scholar] [CrossRef]
- Goula, A.; Gkioka, V.; Michalopoulos, E.; Katsimpoulas, M.; Noutsias, M.; Sarri, E.F.; Stavropoulos, C.; Kostakis, A. Advanced Therapy Medicinal Products Challenges and Perspectives in Regenerative Medicine. J. Clin. Med. Res. 2020, 12, 780–786. [Google Scholar] [CrossRef]
- Cockburn, E.C. EudraLex—The Rules Governing Medicinal Products in the European Union: Volume 4—EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterniary Use; European Union: Brussels, Belgium, 2012. [Google Scholar]
- Pharmacopoiea. 2022. Available online: https://pheur.edqm.eu/home (accessed on 6 March 2022).
- 797 Pharmaceutical Compounding—Sterile Preparations. 2021. Available online: https://doi.usp.org/USPNF/USPNF_M99925_06_01.html (accessed on 6 March 2022).
- Brunner, D. PIC/S Guide to Good Manufacturing Practice for Medicinal Products; PIC/S: Geneva, Switzerland, 2021. [Google Scholar]
- European Medicines Agency. Guideline on the Risk-Based Approach According to Annex I, part IV of Directive 2001/83/EC Applied to Advanced Therapy Medicinal Products; European Medicines Agency: Amsterdam, The Netherlands, 2013.
- Bourne, R.R.A.; Jonas, J.B.; Bron, A.M.; Cicinelli, M.V.; Das, A.; Flaxman, S.R.; Friedman, D.S.; Keeffe, J.E.; Kempen, J.H.; Leasher, J.; et al. Prevalence and causes of vision loss in high-income countries and in Eastern and Central Europe in 2015: Magnitude, temporal trends and projections. Br. J. Ophthalmol. 2018, 102, 575–585. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef]
- Izsvák, Z.; Chuah, M.K.L.; VandenDriessche, T.; Ivics, Z. Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods 2009, 49, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell. Tissue Bank. 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Thumann, G.; Harmening, N.; Prat-Souteyrand, C.; Marie, C.; Pastor, M.; Sebe, A.; Miskey, C.; Hurst, L.D.; Diarra, S.; Kropp, M.; et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017, 6, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Lappas, A.; Weinberger, A.W.A.; Foerster, A.M.H.; Kube, T.; Rezai, K.A.; Kirchhof, B. Iris pigment epithelial cell translocation in exudative age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2000, 238, 631–641. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Monés, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology 2021, 128, 576–586. [Google Scholar] [CrossRef]
- Dreismann, A.K.; McClements, M.E.; Barnard, A.R.; Orhan, E.; Hughes, J.P.; Lachmann, P.J.; MacLaren, R.E. Functional expression of complement factor I following AAV-mediated gene delivery in the retina of mice and human cells. Gene Ther. 2021, 28, 265–276. [Google Scholar] [CrossRef]
- Pastor, M.; Johnen, S.; Harmening, N.; Quiviger, M.; Pailloux, J.; Kropp, M.; Walter, P.; Ivics, Z.; Izsvák, Z.; Thumann, G.; et al. The Antibiotic-free pFAR4 Vector Paired with the Sleeping Beauty Transposon System Mediates Efficient Transgene Delivery in Human Cells. Mol. Ther. Nucleic Acids 2018, 11, 57–67. [Google Scholar] [CrossRef]
- Kropp, M.; Harmening, N.; Bautzová, T.; Asrih, M.; Sealy, G.; Johnen, S.; Izsvák, Z.; Marie, C.; Scherman, D.; van den Berg, J.; et al. Development of GMP-compliant production of freshly isolated and transfected iris pigment epithelial (IPE) cells to treat age-related macular degeneration (AMD). Hum. Gene Ther. 2017, 28, A126. [Google Scholar]
- Bakker, N.A.; Boer, R.; Marie, C.; Scherman, D.; Haanen, J.B.; Beijnen, J.H.; Nuijen, B.; van den Berg, J.H. Small-scale GMP production of plasmid DNA using a simplified and fully disposable production method. J. Biotechnol. X 2019, 2, 100007. [Google Scholar] [CrossRef]
- Harmening, N.; Sealy, G.; Johnen, S.; Kropp, M.; Ronchetti, M.; Aranda, P.; Marie, C.; Scherman, D.; Izsvak, Z.; Thumann, G. Translation of GLP-grade electroporation of primary pigment epithelial cells to GMP-grade GTMP manufacturing for clinical use. Hum. Gene Ther. 2016, 27, A108. [Google Scholar]
- Zanini, C.; Severina, F.; Lando, G.; Fanizza, C.; Cesana, E.; Desidera, D.; Bonifacio, M. Good Design Practices for an integrated containment and production system for Advanced Therapies. Biotechnol. Bioeng. 2020, 117, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Zanini, C. The Concept of Biological Confinement (Bio-Confinement) in Closed Systems for Advanced Therapy Medicinal Products Production. Available online: https://www.linkedin.com/pulse/concept-biological-confinement-bio-confinement-closed-cristina-zanini/ (accessed on 16 September 2020).
- Tack, P.; Victor, J.; Gemmel, P.; Annemans, L. 3D-printing techniques in a medical setting: A systematic literature review. Biomed. Eng. Online 2016, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, J.F.; Hein, A.; Damiani, R. Nuclear DNA Content Varies with Cell Size across Human Cell Types. Cold Spring Harb. Perspect. Biol. 2015, 7, a019091. [Google Scholar] [CrossRef]
- Garcia-Garcia, L.; Recalde, S.; Hernandez, M.; Bezunartea, J.; Rodriguez-Madoz, J.R.; Johnen, S.; Diarra, S.; Marie, C.; Izsvák, Z.; Ivics, Z.; et al. Long-Term PEDF Release in Rat Iris and Retinal Epithelial Cells after Sleeping Beauty Transposon-Mediated Gene Delivery. Mol. Ther. Nucleic Acids 2017, 9, 1–11. [Google Scholar] [CrossRef]
- Bak, A.; Friis, K.P.; Wu, Y.; Ho, R.J.Y. Translating Cell and Gene Biopharmaceutical Products for Health and Market Impact. Product Scaling From Clinical to Marketplace: Lessons Learned and Future Outlook. J. Pharm. Sci. 2019, 108, 3169–3175. [Google Scholar] [CrossRef]
- Ali, N.; Kirchhoff, J.; Onoja, P.I.; Tannert, A.; Neugebauer, U.; Popp, J.; Bocklitz, T. Predictive Modeling of Antibiotic Susceptibility in E. Coli Strains Using the U-Net Network and One-Class Classification. IEEE Access 2020, 8, 167711–167720. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Naldini, L. Toward Clinical Translation of New Gene Targeting Technologies for Correcting Inherited Mutations and Empowering Adoptive Immunotherapy of Cancer (SUPERSIST). Hum. Gene Ther. Clin. Dev. 2015, 26, 95–97. [Google Scholar] [CrossRef]
- Ciulla, T.A.; Hussain, R.M.; Berrocal, A.M.; Nagiel, A. Voretigene neparvovec-rzyl for treatment of RPE65-mediated inherited retinal diseases: A model for ocular gene therapy development. Expert Opin. Biol. Ther. 2020, 20, 565–578. [Google Scholar] [CrossRef]
- Gupta, V.; Lourenço, S.P.; Hidalgo, I.J. Development of Gene Therapy Vectors: Remaining Challenges. J. Pharm. Sci. 2021, 110, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar] [CrossRef] [PubMed]
- EMA (European Medicines Agency). Guidelines Relevant for Advanced Therapy Medicinal Products. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/advanced-therapies/guidelines-relevant-advanced-therapy-medicinal-products (accessed on 6 March 2022).
- Krishna, A.K.; Lodhi, S.A.; Harris, M.R. Isolation technology for research and development applications: From concept to production. Pharm. Dev. Technol. 2000, 5, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Kropp, M.; Tian, S.; Harmening, N.; Johnen, S.; Scherman, D.; Marie, C.; Izsvák, Z.; Thumann, G. Over-expression of PEDF by PEDF-transfected primary pigment epithelial cells does not induce tumorigenicity. Hum. Gene Ther. 2015, 26, A81. [Google Scholar]
- Tombran-Tink, J. The Neuroprotective and Angiogenesis Inhibitory Serpin, PEDF: New Insights into Phylogeny, Function, and Signaling. Front. Biosci. 2005, 10, 2131–2149. [Google Scholar] [CrossRef]
- Schott, J.W.; Jaeschke, N.M.; Hoffmann, D.; Maetzig, T.; Ballmaier, M.; Godinho, T.; Cathomen, T.; Schambach, A. Deciphering the impact of parameters influencing transgene expression kinetics after repeated cell transduction with integration-deficient retroviral vectors. Cytometry A 2015, 87, 405–418. [Google Scholar] [CrossRef]
- Herweijer, H.; Zhang, G.; Subbotin, V.M.; Budker, V.; Williams, P.; Wolff, J.A. Time course of gene expression after plasmid DNA gene transfer to the liver. J. Gene Med. 2001, 3, 280–291. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal No. | No. Examinations | Δ Weight (%) | Behavior | Food/Water Intake | Fur | Blinking | Eyes |
|---|---|---|---|---|---|---|---|
| 1 | 11 | 0 | N | N | N | N | N |
| 2 | 11 | −2.6 | N | N | N | N | N |
| 3 | 2 | −2.2 | N | N | N | N | N |
| 4 | 2 | −11.1 | N | N | N | N | N |
| Animal No. | Fundus Morphology | IOP Treated Eye (mm/Hg) | IOP Contralateral Eye (mm/Hg) | p-Value |
|---|---|---|---|---|
| 1 | Normal | 13 (day 0) | 14 (day 0) | p > 0.999 |
| 8 (day 7) | 10 (day 7) | |||
| 7 (day 90) | 8 (day 90) | |||
| 2 | Normal | 14 (day 0) | 12 (day 0) | p = 0.580 |
| 6 (day 7) | 8 (day 7) | |||
| 9 (day 90) | 10 (day 90) |
| Total (Mean ± SD) | Training (Mean ± SD) | ValRun (Mean ± SD) | |
|---|---|---|---|
| No. of donors | 28 | 11 | 17 |
| No. of eyes | 42 | 16 | 26 |
| No. of productions | 143 | 39 | 104 |
| Age of donors (years) | 81.18 ± 11.73 | 78.09 ± 15.46 | 83.65 ± 8.93 |
| Sex of donors (m/f) | 12/16 | 3/8 | 8/9 |
| Time of death to cell isolation (d) | 7.24 ± 1.23 | 7.45 ± 0.82 | 7.35 ± 1.29 |
| Release Criterion/Phase | Method for Its Determination | Specification | Action (In Case of a Result OOS) | |
|---|---|---|---|---|
| Validation Run | Clinical Trial | |||
| Starting material (iridectomy) | Visual control: no saliencies | 0 | Stop production | Stop production |
| CliniporatorTM | Self-test | “Passed“ | Repetition of self-test | Repetition of self-test |
| 2nd self-test OOS: stop production | 2nd self-test OOS: stop production | |||
| Plasmidmixture | Visual control: vial intact & frozen | “Yes” | New vial | New vial |
| No conforming vial available: continue production | No conforming vial available: stop production | |||
| 3P.14 buffer | Visual control: vial intact & frozen | “Yes” | New vial | New vial |
| No conforming vial available: continue production | No conforming vial available: stop production | |||
| Transport | Visual control: tape intact | “Yes” | Continue production; record time & temperature | Stop production |
| Starting material (stroma) | Eur. Ph. Method 11.0, 2.6.: microbiology (stroma) | 0 CFU | N.A. | Antibiotic regimen; not critical for batch release |
| Homogenization | Visual control: homogenous suspension | “Yes” | Repeat resuspension | Repeat resuspension |
| Volume | Visual control: 15 µL | <15 µL | Add 3P.14 buffer up to 15 µL | Add 3P.14 buffer up to 15 µL |
| Electroporation | Normal performance | No error message | Continue production | Stop production |
| Washing (centrifugation) | Divide supernatant into 3 parts: | |||
| tIPE | Eur. Ph. Method 11.0, 2.6.: microbiology | 0 CFU | N.A. | Antibiotic regimen; not critical for batch release |
| Bacterial endotoxin | EndoSafe®, colorimetric | <0.2 EU/mL | Continue production | Stop production |
| Remnant extracellular DNA | Qubit dsDNA assay, fluorimetric | ≥33% | N.A. | N.A. (not measured) |
| Cell number | Neubauer chamber: counting | ≥5000 cells | Continue production | Continue production; open a deviation |
| Cell viability | Percentage viable cells | tIPE ≈ Co-1/2 | N.A. | N.A. (not measured) |
| Filling syringe | Visual control: absence of bubbles | <1 | Repeat filling up to 3 times | Repeat filling up to 3 times |
| Visual control: volume | ≥15 µL/33% | 3rd try failed: continue production | 3rd try failed: stop production | |
| Duration | Timer: time | ≤30 min | Continue production | Continue production; open a deviation |
| tIPE labelling | Completeness and readability | “Yes” | Correct labelling | Correct labelling |
| Air samples, settle plates, contact plates, glove prints | Eur. Ph. Method 11.0, 2.6.: microbiology | 0 CFU | N.A. | Antibiotic regimen; not critical for batch release. |
| Particle control | Logiview and Labguard®-controlled: inorganic particles | Class A 0.5 µm/m3: <3520 5 µm/m3: <29 Class C 0.5 µm/m3: <3.5 Mio 5 µm/m3: <29,300 | Continue production | Stop production |
| Dossier | 3-fold control: completeness, readability, correctness | “Yes” | Correction | Correction |
| No correction possible: documentation of non-compliant production | No correction possible: no release | |||
| P1 | P2 | P3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N (%) | Value (Mean ± SD) | Conforming (%) | N (%) | Value (Mean ± SD) | Conforming (%) | N (%) | Value (Mean ± SD) | Conforming (%) | |
| Productions | |||||||||
| Total | 40 | 24 | 40 | ||||||
| Valid | 35 | 19 | 34 | ||||||
| Compliant | 23 (65.7) | 10 (52.6) | 19 (55.9) | ||||||
| Consecutive compliant | 7 | 0 | 5 | ||||||
| Cells | |||||||||
| No. | 40 | 18,060 ± 9291 | 38 (95.0) | 24 | 17,448 ± 15,824 | 23 (95.8) | 40 | 18,238 ± 10,948 | 39 (97.5) |
| Viability (%) | 28 | 31.76 ± 20.65 | n.a. | 12 | 28.9 ± 18.7 | n.a. | 28 | 33.4 ± 18.02 | n.a. |
| Remnant extracellular DNA (%) | 33 | 82.74 ± 22.98 | 26 (78.8) | 20 | 74.8 ± 24.2 | 15 (75.0) | 34 | 80.9 ± 19.3 | 32 (94.1) |
| Time (min) | 39 | 19.1 ± 2.15 | 39 (100) | 24 | 19.88 ± 2.21 | 24 (100) | 40 | 20.12 ± 2.78 | 40 (100) |
| tIPE volume (µL) | 40 | 79.7 ± 11.2 | 40 (100) | 24 | 83.4 ± 12.1 | 24 (100) | 40 | 81.3 ± 13.7 | 40 (100) |
| Endotoxins | 40 | n.a. | 39 (97.5) | 24 | n.a. | 24 (100) | 39 | n.a. | 33 (84.6) |
| Iridectomy sterility | 40 | n.a. | 38 (95.0) | 24 | n.a. | 23 (95.8) | 40 | n.a. | 39 (97.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kropp, M.; Harmening, N.; Bascuas, T.; Johnen, S.; De Clerck, E.; Fernández, V.; Ronchetti, M.; Cadossi, R.; Zanini, C.; Scherman, D.; et al. GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration. Biomedicines 2022, 10, 2777. https://doi.org/10.3390/biomedicines10112777
Kropp M, Harmening N, Bascuas T, Johnen S, De Clerck E, Fernández V, Ronchetti M, Cadossi R, Zanini C, Scherman D, et al. GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration. Biomedicines. 2022; 10(11):2777. https://doi.org/10.3390/biomedicines10112777
Chicago/Turabian StyleKropp, Martina, Nina Harmening, Thais Bascuas, Sandra Johnen, Eline De Clerck, Verónica Fernández, Mattia Ronchetti, Ruggero Cadossi, Cristina Zanini, Daniel Scherman, and et al. 2022. "GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration" Biomedicines 10, no. 11: 2777. https://doi.org/10.3390/biomedicines10112777
APA StyleKropp, M., Harmening, N., Bascuas, T., Johnen, S., De Clerck, E., Fernández, V., Ronchetti, M., Cadossi, R., Zanini, C., Scherman, D., Ivics, Z., Marie, C., Izsvák, Z., & Thumann, G. (2022). GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration. Biomedicines, 10(11), 2777. https://doi.org/10.3390/biomedicines10112777