
Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
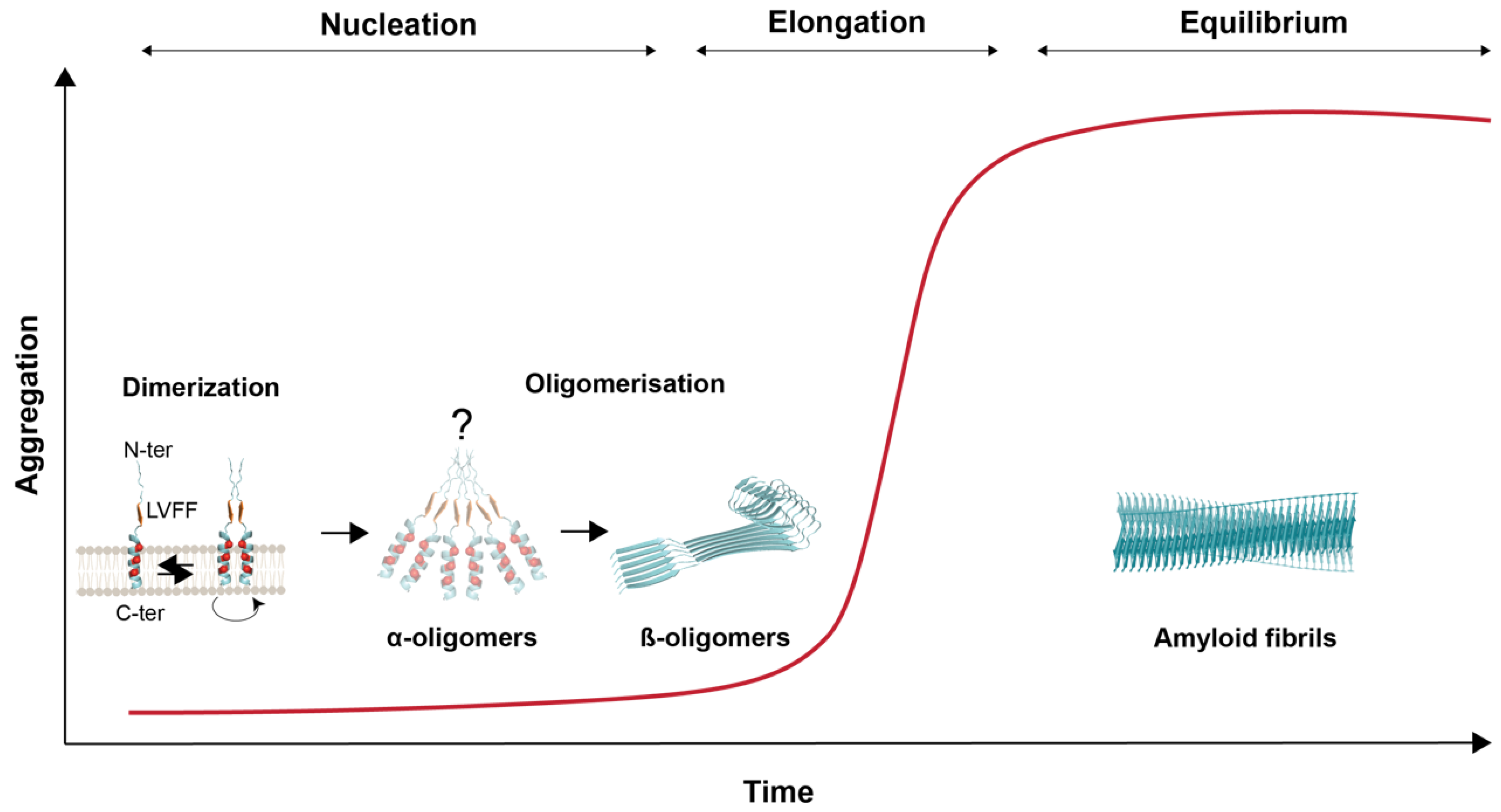{kind=link}
Abstract
1. Introduction
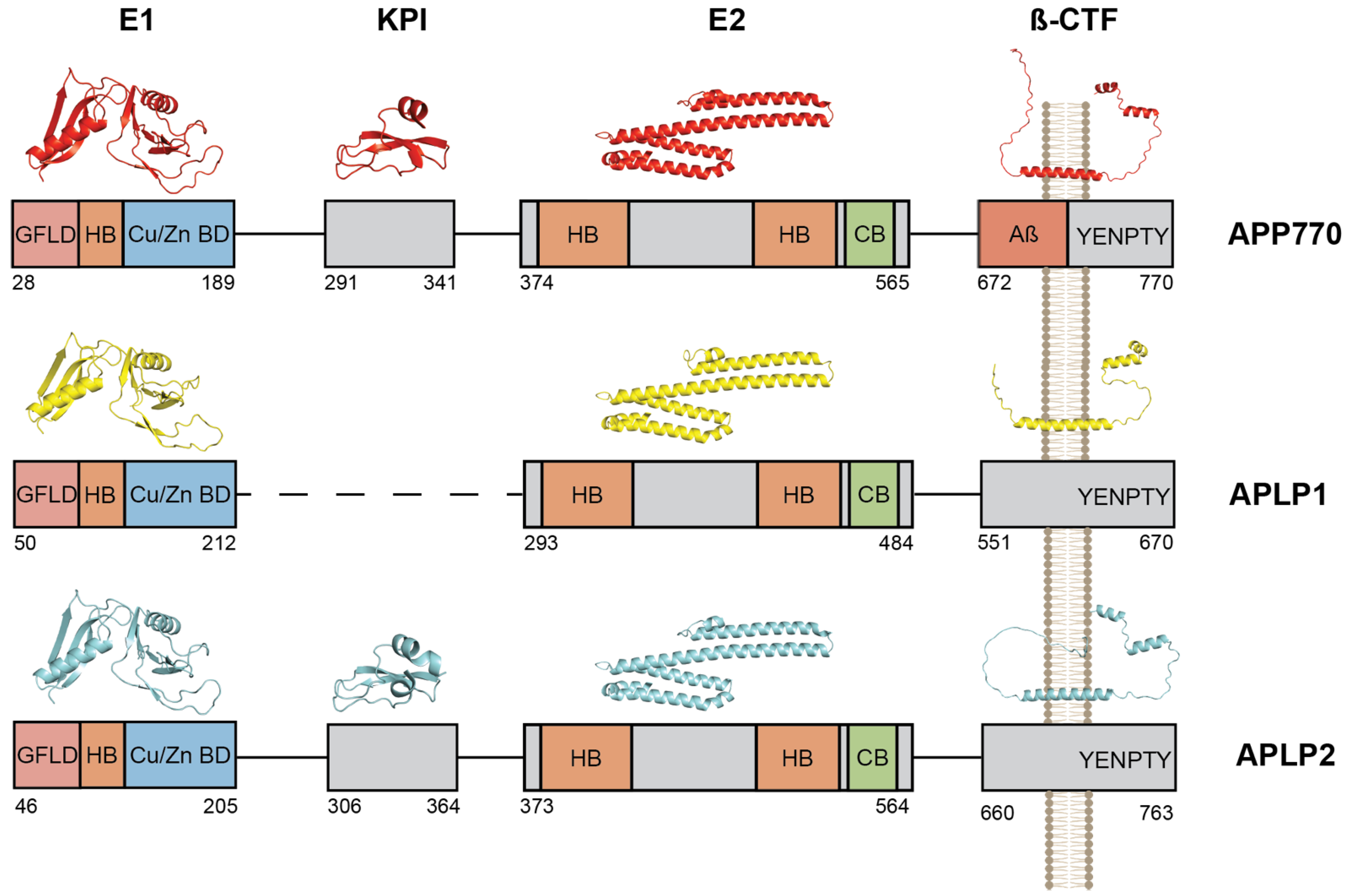
2. The Amyloid Precursor Protein Family and Aβ Production
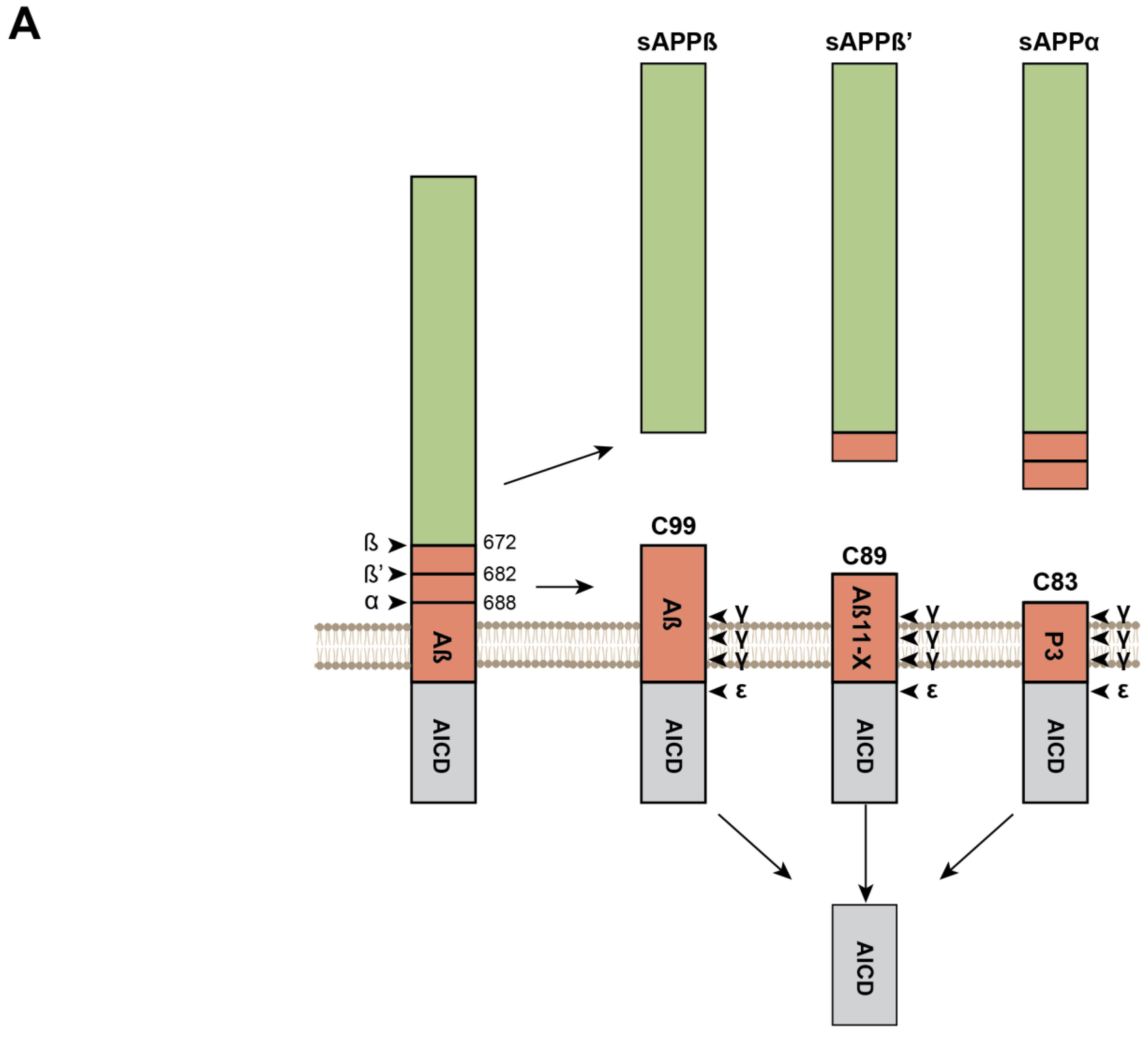
Sequential Processing of the APP and APLPs and Production of β-Amyloid Peptides
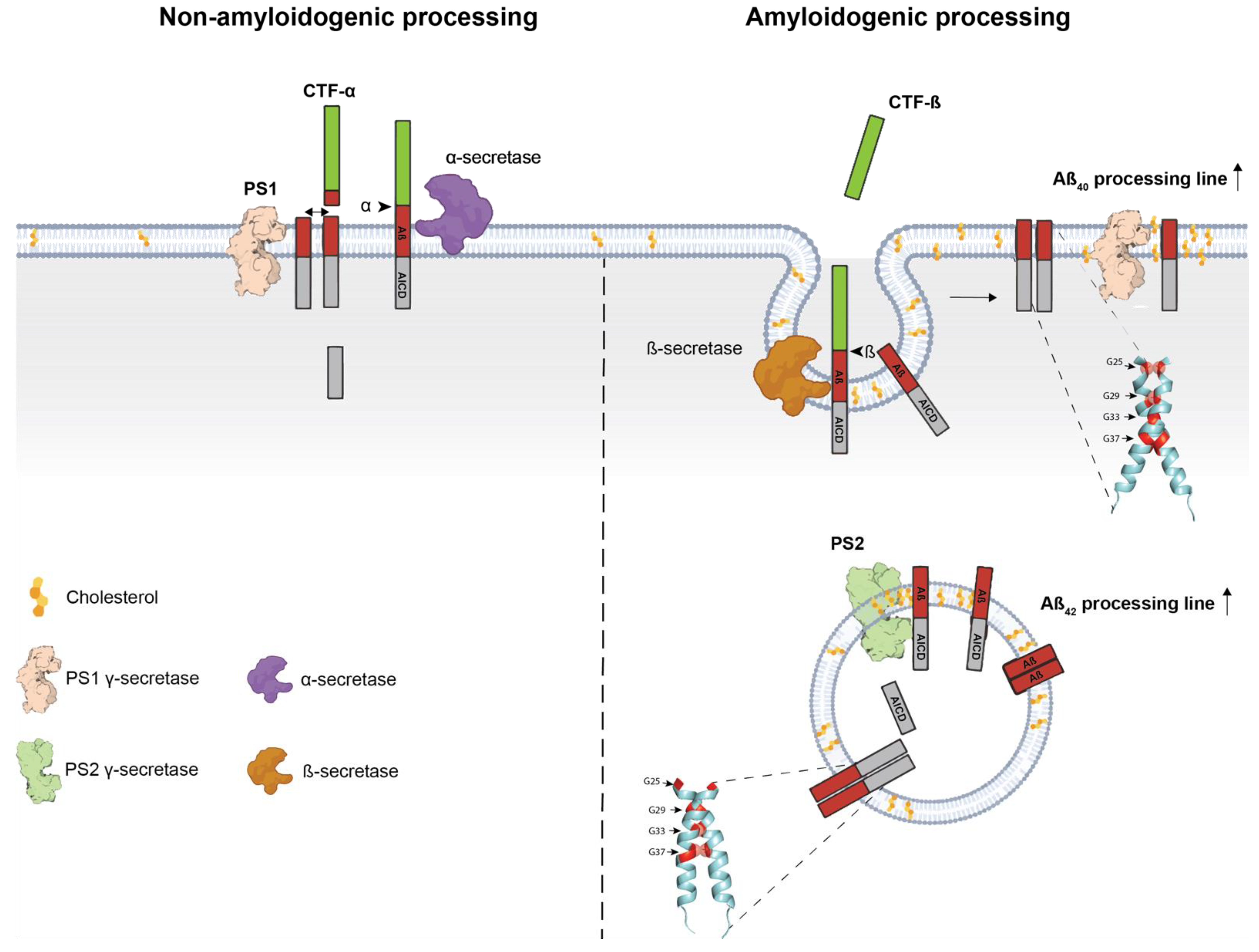
3. Transmembrane Interactions and Amyloidogenic Processing

3.1. Dimeric Conformation of C99 Regulates γ-Secretase Processing
3.2. APP Dimeric Conformation Controls Its Intracellular Localization and Aβ Generation
3.3. The Role of Lipids in C99 Dimerization and Amyloidogenic Processing
3.4. Impact of Familial AD Mutations on APP TM Dimerization: A Link with Aβ Production?
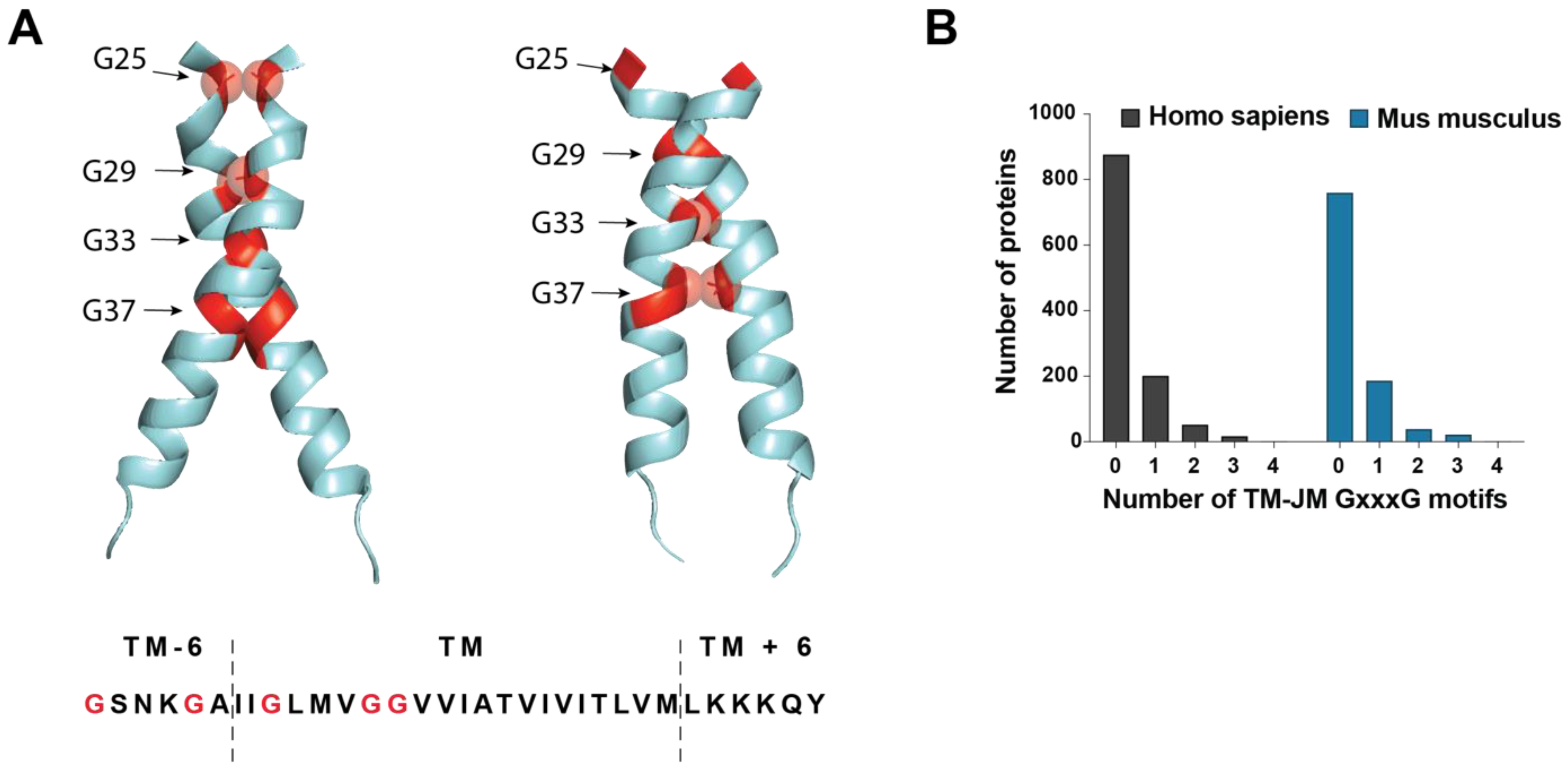
3.5. JM/TM Determinants Drive Aggregation Steps (Nucleation) Leading to Pathological Aβ Seeds

4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Walker, L.C.; LeVine, H. The cerebral proteopathies. Neurobiol. Aging 2000, 21, 559–561. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI–Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Goto, Y.; Noji, M.; Nakajima, K.; Yamaguchi, K. Supersaturation-Dependent Formation of Amyloid Fibrils. Molecules 2022, 27, 4588. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S33–S38. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Hardy, J. Alzheimer’s disease: The amyloid cascade hypothesis: An update and reappraisal. J. Alzheimers. Dis 2006, 9, 151–153. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef]
- Slunt, H.H.; Thinakaran, G.; Von Koch, C.; Lo, A.C.; Tanzi, R.E.; Sisodia, S.S. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J. Biol. Chem. 1994, 269, 2637–2644. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.; St George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Wasco, W.; Bupp, K.; Magendantz, M.; Gusella, J.F.; Tanzi, R.E.; Solomon, F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10758–10762. [Google Scholar] [CrossRef]
- Musa, A.; Lehrach, H.; Russo, V.A. Distinct expression patterns of two zebrafish homologues of the human APP gene during embryonic development. Dev. Genes Evol. 2001, 211, 563–567. [Google Scholar] [CrossRef]
- Rosen, D.R.; Martin-Morris, L.; Luo, L.Q.; White, K. A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1989, 86, 2478–2482. [Google Scholar] [CrossRef]
- Shariati, S.A.; De Strooper, B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013, 587, 2036–2045. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakamura, S.; Ueda, K.; Kameyama, M.; Shiojiri, S.; Takahashi, Y.; Kitaguchi, N.; Ito, H. Three types of amyloid protein precursor mRNA in human brain: Their differential expression in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1988, 157, 472–479. [Google Scholar] [CrossRef]
- Sprecher, C.A.; Grant, F.J.; Grimm, G.; O'Hara, P.J.; Norris, F.; Norris, K.; Foster, D.C. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: Evidence for a multigene family. Biochemistry 1993, 32, 4481–4486. [Google Scholar] [CrossRef]
- Wasco, W.; Gurubhagavatula, S.; Paradis, M.D.; Romano, D.M.; Sisodia, S.S.; Hyman, B.T.; Neve, R.L.; Tanzi, R.E. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat. Genet. 1993, 5, 95–100. [Google Scholar] [CrossRef]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef]
- Wang, Y.; Ha, Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol. Cell 2004, 15, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xue, Y.; Hu, J.; Wang, Y.; Liu, X.; Demeler, B.; Ha, Y. The E2 Domains of APP and APLP1 Share a Conserved Mode of Dimerization. Biochemistry 2011, 50, 5453–5464. [Google Scholar] [CrossRef]
- Roisman, L.C.; Han, S.; Chuei, M.J.; Connor, A.R.; Cappai, R. The crystal structure of amyloid precursor-like protein 2 E2 domain completes the amyloid precursor protein family. FASEB J. 2019, 33, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Hoefgen, S.; Roeser, D.; Schlott, B.; Gührs, K.H.; Than, M.E. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2010, 107, 5381–5386. [Google Scholar] [CrossRef]
- Hynes, T.R.; Randal, M.; Kennedy, L.A.; Eigenbrot, C.; Kossiakoff, A.A. X-ray crystal structure of the protease inhibitor domain of Alzheimer’s amyloid beta-protein precursor. Biochemistry 1990, 29, 10018–10022. [Google Scholar] [CrossRef]
- Beher, D.; Hesse, L.; Masters, C.L.; Multhaup, G. Regulation of Amyloid Protein Precursor (APP) Binding to Collagen and Mapping of the Binding Sites on APP and Collagen Type I (∗). J. Biol. Chem. 1996, 271, 1613–1620. [Google Scholar] [CrossRef]
- Schubert, D.; LaCorbiere, M.; Saitoh, T.; Cole, G. Characterization of an amyloid beta precursor protein that binds heparin and contains tyrosine sulfate. Proc. Natl. Acad. Sci. USA 1989, 86, 2066–2069. [Google Scholar] [CrossRef]
- Small, D.H.; Nurcombe, V.; Reed, G.; Clarris, H.; Moir, R.; Beyreuther, K.; Masters, C.L. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J. Neurosci. 1994, 14, 2117–2127. [Google Scholar] [CrossRef]
- Hoefgen, S.; Coburger, I.; Roeser, D.; Schaub, Y.; Dahms, S.O.; Than, M.E. Heparin induced dimerization of APP is primarily mediated by E1 and regulated by its acidic domain. J. Struct. Biol. 2014, 187, 30–37. [Google Scholar] [CrossRef][Green Version]
- Soba, P.; Eggert, S.; Wagner, K.; Zentgraf, H.; Siehl, K.; Kreger, S.; Löwer, A.; Langer, A.; Merdes, G.; Paro, R.; et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005, 24, 3624–3634. [Google Scholar] [CrossRef]
- Bush, A.I.; Multhaup, G.; Moir, R.D.; Williamson, T.G.; Small, D.H.; Rumble, B.; Pollwein, P.; Beyreuther, K.; Masters, C.L. A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 1993, 268, 16109–16112. [Google Scholar] [CrossRef]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Mayer, M.C.; Kaden, D.; Schauenburg, L.; Hancock, M.A.; Voigt, P.; Roeser, D.; Barucker, C.; Than, M.E.; Schaefer, M.; Multhaup, G. Novel zinc-binding site in the E2 domain regulates amyloid precursor-like protein 1 (APLP1) oligomerization. J. Biol. Chem. 2014, 289, 19019–19030. [Google Scholar] [CrossRef] [PubMed]
- Dienemann, C.; Coburger, I.; Mehmedbasic, A.; Andersen, O.M.; Than, M.E. Mutants of metal binding site M1 in APP E2 show metal specific differences in binding of heparin but not of sorLA. Biochemistry 2015, 54, 2490–2499. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Könnig, I.; Roeser, D.; Gührs, K.H.; Mayer, M.C.; Kaden, D.; Multhaup, G.; Than, M.E. Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 2012, 416, 438–452. [Google Scholar] [CrossRef]
- Noda, Y.; Asada, M.; Kubota, M.; Maesako, M.; Watanabe, K.; Uemura, M.; Kihara, T.; Shimohama, S.; Takahashi, R.; Kinoshita, A.; et al. Copper enhances APP dimerization and promotes Aβ production. Neurosci. Lett. 2013, 547, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Rohan de Silva, H.A.; Jen, A.; Wickenden, C.; Jen, L.S.; Wilkinson, S.L.; Patel, A.J. Cell-specific expression of beta-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Mol. Brain Res. 1997, 47, 147–156. [Google Scholar] [CrossRef]
- Mahdi, F.; Van Nostrand, W.E.; Schmaier, A.H. Protease nexin-2/amyloid beta-protein precursor inhibits factor Xa in the prothrombinase complex. J. Biol. Chem. 1995, 270, 23468–23474. [Google Scholar] [CrossRef]
- Scheinfeld, M.H.; Ghersi, E.; Laky, K.; Fowlkes, B.J.; D’Adamio, L. Processing of beta-amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J. Biol. Chem. 2002, 277, 44195–44201. [Google Scholar] [CrossRef] [PubMed]
- Beckett, C.; Nalivaeva, N.N.; Belyaev, N.D.; Turner, A.J. Nuclear signalling by membrane protein intracellular domains: The AICD enigma. Cell Signal. 2012, 24, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Pardossi-Piquard, R.; Checler, F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2012, 120 (Suppl. S1), 109–124. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Sudhof, T.C. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Sudhof, T.C. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J. Biol. Chem 2004, 279, 24601–24611. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; Serneels, L.; Tolia, A.; Craessaerts, K.; Derks, C.; Filippov, M.A.; Muller, U.; De Strooper, B. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006, 7, 739–745. [Google Scholar] [CrossRef]
- Opsomer, R.; Contino, S.; Perrin, F.; Gualdani, R.; Tasiaux, B.; Doyen, P.; Vergouts, M.; Vrancx, C.; Doshina, A.; Pierrot, N.; et al. Amyloid Precursor Protein (APP) Controls the Expression of the Transcriptional Activator Neuronal PAS Domain Protein 4 (NPAS4) and Synaptic GABA Release. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Sisodia, S.S.; Koo, E.H.; Beyreuther, K.; Unterbeck, A.; Price, D.L. Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495. [Google Scholar] [CrossRef]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124. [Google Scholar] [CrossRef]
- Eggert, S.; Paliga, K.; Soba, P.; Evin, G.; Masters, C.L.; Weidemann, A.; Beyreuther, K. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: Modulation of APLP-1 processing by n-glycosylation. J. Biol. Chem. 2004, 279, 18146–18156. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. Embo J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Adlerz, L.; Multhaup, G.; Iverfeldt, K. Insulin-like Growth Factor-1 (IGF-1)-induced Processing of Amyloid-β Precursor Protein (APP) and APP-like Protein 2 Is Mediated by Different Metalloproteinases*. J. Biol. Chem. 2010, 285, 10223–10231. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA 1992, 89, 6075–6079. [Google Scholar] [CrossRef]
- Dislich, B.; Lichtenthaler, S.F. The Membrane-Bound Aspartyl Protease BACE1: Molecular and Functional Properties in Alzheimer’s Disease and Beyond. Front Physiol. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.; Wang, R.; Zhang, X.; Zhang, S.; Wu, Y.; Staufenbiel, M.; Cai, F.; Song, W. Amyloid-β protein (Aβ) Glu11 is the major β-secretase site of β-site amyloid-β precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Aβ Asp1 contributes to Alzheimer pathogenesis. Eur. J. Neurosci. 2013, 37, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Z.; Cai, F.; Zhang, M.; Wu, Y.; Zhang, J.; Song, W. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J. Neurosci. 2017, 37, 6915–6925. [Google Scholar] [CrossRef]
- Kimura, A.; Hata, S.; Suzuki, T. Alternative Selection of β-Site APP-Cleaving Enzyme 1 (BACE1) Cleavage Sites in Amyloid β-Protein Precursor (APP) Harboring Protective and Pathogenic Mutations within the Aβ Sequence. J. Biol. Chem. 2016, 291, 24041–24053. [Google Scholar] [CrossRef]
- Weidemann, A.; Eggert, S.; Reinhard, F.B.; Vogel, M.; Paliga, K.; Baier, G.; Masters, C.L.; Beyreuther, K.; Evin, G. A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 2002, 41, 2825–2835. [Google Scholar] [CrossRef]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive Tripeptide and Tetrapeptide Release from the Transmembrane Domain of β-Carboxyl Terminal Fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase. Elife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Hogl, S.; Kuhn, P.H.; Colombo, A.; Lichtenthaler, S.F. Determination of the proteolytic cleavage sites of the amyloid precursor-like protein 2 by the proteases ADAM10, BACE1 and γ-secretase. PLoS ONE 2011, 6, e21337. [Google Scholar] [CrossRef]
- Liu, R.; McAllister, C.; Lyubchenko, Y.; Sierks, M.R. Residues 17-20 and 30-35 of beta-amyloid play critical roles in aggregation. J. Neurosci. Res. 2004, 75, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Kienlen-Campard, P.; Tang, T.-C.; Perrin, F.; Opsomer, R.; Decock, M.; Pan, X.; Octave, J.-N.; Constantinescu, S.N.; Smith, S.O. β-Sheet Structure within the Extracellular Domain of C99 Regulates Amyloidogenic Processing. Sci. Rep. 2017, 7, 17159. [Google Scholar] [CrossRef] [PubMed]
- Söldner, C.A.; Sticht, H.; Horn, A.H.C. Role of the N-terminus for the stability of an amyloid-β fibril with three-fold symmetry. PLoS ONE 2017, 12, e0186347. [Google Scholar] [CrossRef]
- Scheuermann, S.; Hambsch, B.; Hesse, L.; Stumm, J.; Schmidt, C.; Beher, D.; Bayer, T.A.; Beyreuther, K.; Multhaup, G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J. Biol. Chem. 2001, 276, 33923–33929. [Google Scholar] [CrossRef] [PubMed]
- Ben Khalifa, N.; Tyteca, D.; Marinangeli, C.; Depuydt, M.; Collet, J.F.; Courtoy, P.J.; Renauld, J.C.; Constantinescu, S.; Octave, J.N.; Kienlen-Campard, P. Structural features of the KPI domain control APP dimerization, trafficking, and processing. FASEB J. 2012, 26, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Kienlen-Campard, P.; Tasiaux, B.; Van Hees, J.; Li, M.; Huysseune, S.; Sato, T.; Fei, J.Z.; Aimoto, S.; Courtoy, P.J.; Smith, S.O.; et al. Amyloidogenic Processing but Not Amyloid Precursor Protein (APP) Intracellular C-terminal Domain Production Requires a Precisely Oriented APP Dimer Assembled by Transmembrane GXXXG Motifs. J. Biol. Chem. 2008, 283, 7733–7744. [Google Scholar] [CrossRef] [PubMed]
- Munter, L.M.; Voigt, P.; Harmeier, A.; Kaden, D.; Gottschalk, K.E.; Weise, C.; Pipkorn, R.; Schaefer, M.; Langosch, D.; Multhaup, G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007, 26, 1702–1712. [Google Scholar] [CrossRef]
- Adams, P.D.; Engelman, D.M.; Brunger, A.T. Improved prediction for the structure of the dimeric transmembrane domain of glycophorin A obtained through global searching. Proteins 1996, 26, 257–261. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Treutlein, H.R.; Adams, P.D.; Brunger, A.T.; Engelman, D.M. A dimerization motif for transmembrane alpha-helices. Nat. Struct. Biol. 1994, 1, 157–163. [Google Scholar] [CrossRef]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol 2000, 296, 911–919. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Flanagan, J.M.; Treutlein, H.R.; Zhang, J.; Engelman, D.M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 1992, 31, 12719–12725. [Google Scholar] [CrossRef] [PubMed]
- Teese, M.G.; Langosch, D. Role of GxxxG Motifs in Transmembrane Domain Interactions. Biochemistry 2015, 54, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Kirrbach, J.; Krugliak, M.; Ried, C.L.; Pagel, P.; Arkin, I.T.; Langosch, D. Self-interaction of transmembrane helices representing pre-clusters from the human single-span membrane proteins. Bioinformatics 2013, 29, 1623–1630. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hendriks, L.; van Duijn, C.M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M.G.; Antonarakis, S.E.; Martin, J.J.; et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef]
- Harrison, C.F.; Lawson, V.A.; Coleman, B.M.; Kim, Y.S.; Masters, C.L.; Cappai, R.; Barnham, K.J.; Hill, A.F. Conservation of a glycine-rich region in the prion protein is required for uptake of prion infectivity. J. Biol. Chem. 2010, 285, 20213–20223. [Google Scholar] [CrossRef]
- Tang, T.-C.; Hu, Y.; Kienlen-Campard, P.; El Haylani, L.; Decock, M.; Van Hees, J.; Fu, Z.; Octave, J.-N.; Constantinescu, S.N.; Smith, S.O. Conformational changes induced by the A21G Flemish mutation in the amyloid precursor protein lead to increased Aβ production. Structure 2014, 22, 387–396. [Google Scholar] [CrossRef]
- Beel, A.J.; Mobley, C.K.; Kim, H.J.; Tian, F.; Hadziselimovic, A.; Jap, B.; Prestegard, J.H.; Sanders, C.R. Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): Does APP function as a cholesterol sensor? Biochemistry 2008, 47, 9428–9446. [Google Scholar] [CrossRef]
- Nadezhdin, K.D.; Bocharova, O.V.; Bocharov, E.V.; Arseniev, A.S. Dimeric structure of transmembrane domain of amyloid precursor protein in micellar environment. FEBS Lett. 2012, 586, 1687–1692. [Google Scholar] [CrossRef]
- Perrin, F.; Papadopoulos, N.; Suelves, N.; Opsomer, R.; Vadukul, D.M.; Vrancx, C.; Smith, S.O.; Vertommen, D.; Kienlen-Campard, P.; Constantinescu, S.N. Dimeric Transmembrane Orientations of APP/C99 Regulate γ-Secretase Processing Line Impacting Signaling and Oligomerization. iScience 2020, 23, 101887. [Google Scholar] [CrossRef]
- Decock, M.; El, H.L.; Stanga, S.; Dewachter, I.; Octave, J.N.; Smith, S.O.; Constantinescu, S.N.; Kienlen-Campard, P. Analysis by a highly sensitive split luciferase assay of the regions involved in APP dimerization and its impact on processing. FEBS Open. Biol. 2015, 5, 763–773. [Google Scholar] [CrossRef]
- Decock, M.; Stanga, S.; Octave, J.N.; Dewachter, I.; Smith, S.O.; Constantinescu, S.N.; Kienlen-Campard, P. Glycines from the APP GXXXG/GXXXA Transmembrane Motifs Promote Formation of Pathogenic Abeta Oligomers in Cells. Front. Aging Neurosci. 2016, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Higashide, H.; Ishihara, S.; Nobuhara, M.; Ihara, Y.; Funamoto, S. Alanine substitutions in the GXXXG motif alter C99 cleavage by gamma-secretase but not its dimerization. J. Neurochem. 2017, 140, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Munter, L.M.; Botev, A.; Richter, L.; Hildebrand, P.W.; Althoff, V.; Weise, C.; Kaden, D.; Multhaup, G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J. Biol. Chem 2010, 285, 21636–21643. [Google Scholar] [CrossRef] [PubMed]
- Sagi, S.A.; Lessard, C.B.; Winden, K.D.; Maruyama, H.; Koo, J.C.; Weggen, S.; Kukar, T.L.; Golde, T.E.; Koo, E.H. Substrate sequence influences gamma-secretase modulator activity, role of the transmembrane domain of the amyloid precursor protein. J. Biol. Chem. 2011, 286, 39794–39803. [Google Scholar] [CrossRef]
- Sato, T.; Tang, T.C.; Reubins, G.; Fei, J.Z.; Fujimoto, T.; Kienlen-Campard, P.; Constantinescu, S.N.; Octave, J.N.; Aimoto, S.; Smith, S.O. A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. USA 2009, 106, 1421–1426. [Google Scholar] [CrossRef]
- So, P.P.; Khodr, C.E.; Chen, C.D.; Abraham, C.R. Comparable dimerization found in wildtype and familial Alzheimer’s disease amyloid precursor protein mutants. Am. J. Neurodegener. Dis. 2013, 2, 15–28. [Google Scholar]
- Richter, L.; Munter, L.M.; Ness, J.; Hildebrand, P.W.; Dasari, M.; Unterreitmeier, S.; Bulic, B.; Beyermann, M.; Gust, R.; Reif, B.; et al. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc. Natl. Acad. Sci. USA 2010, 107, 14597–14602. [Google Scholar] [CrossRef]
- Eggert, S.; Midthune, B.; Cottrell, B.; Koo, E.H. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J. Biol. Chem. 2009, 284, 28943–28952. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363. [Google Scholar] [CrossRef]
- Eggert, S.; Gonzalez, A.C.; Thomas, C.; Schilling, S.; Schwarz, S.M.; Tischer, C.; Adam, V.; Strecker, P.; Schmidt, V.; Willnow, T.E.; et al. Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cell. Mol. Life Sci. 2018, 75, 301–322. [Google Scholar] [CrossRef]
- Wong, C.-O. Endosomal-Lysosomal Processing of Neurodegeneration-Associated Proteins in Astrocytes. Int. J. Mol. Sci. 2020, 21, 5149. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, M.; Cai, F.; Song, W. Biological function of Presenilin and its role in AD pathogenesis. Transl. Neurodegener. 2013, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef]
- Bentahir, M.; Nyabi, O.; Verhamme, J.; Tolia, A.; Horré, K.; Wiltfang, J.; Esselmann, H.; De Strooper, B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 2006, 96, 732–742. [Google Scholar] [CrossRef]
- Vrancx, C.; Vadukul, D.M.; Suelves, N.; Contino, S.; D’Auria, L.; Perrin, F.; van Pesch, V.; Hanseeuw, B.; Quinton, L.; Kienlen-Campard, P. Mechanism of Cellular Formation and In Vivo Seeding Effects of Hexameric beta-Amyloid Assemblies. Mol. Neurobiol. 2021, 58, 6647–6669. [Google Scholar] [CrossRef]
- Ye, X.; Cai, Q. Snapin-mediated BACE1 retrograde transport is essential for its degradation in lysosomes and regulation of APP processing in neurons. Cell Rep. 2014, 6, 24–31. [Google Scholar] [CrossRef]
- Huse, J.T.; Pijak, D.S.; Leslie, G.J.; Lee, V.M.; Doms, R.W. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J. Biol. Chem. 2000, 275, 33729–33737. [Google Scholar] [CrossRef]
- Pastorino, L.; Ikin, A.F.; Nairn, A.C.; Pursnani, A.; Buxbaum, J.D. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta). Mol. Cell Neurosci. 2002, 19, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Scott, D.A.; Ganguly, A.; Koo, E.H.; Tang, Y.; Roy, S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 2013, 79, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.; Tasiaux, B.; Opsomer, R.; Hage, S.; Sodero, A.O.; Dewachter, I.; Octave, J.N.; Smith, S.O.; Constantinescu, S.N.; Kienlen-Campard, P. Presenilin transmembrane domain 8 conserved AXXXAXXXG motifs are required for the activity of the gamma-secretase complex. J. Biol. Chem. 2015, 290, 7169–7184. [Google Scholar] [CrossRef]
- Lee, S.F.; Shah, S.; Yu, C.; Wigley, W.C.; Li, H.; Lim, M.; Pedersen, K.; Han, W.; Thomas, P.; Lundkvist, J.; et al. A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J. Biol. Chem. 2004, 279, 4144–4152. [Google Scholar] [PubMed]
- Liu, L.; Lauro, B.M.; Wolfe, M.S.; Selkoe, D.J. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate gamma-secretase function in generating Alzheimer-causing Abeta peptides. J. Biol. Chem. 2021, 296, 100393. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A. über eigenartige Krankheitsfälle des späteren Alters. Z. Für Die Gesamte Neurol. Und Psychiatr. 1911, 4, 356. [Google Scholar] [CrossRef]
- Foley, P. Lipids in Alzheimer’s disease: A century-old story. Biochim. Biophys. Acta 2010, 1801, 750–753. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in Amyloid-β Processing, Aggregation, and Toxicity. In Lipids in Protein Misfolding; Gursky, O., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 67–94. [Google Scholar]
- Grimm, M.O.W.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Emmerling, M.R.; Bisgaier, C.L.; Essenburg, A.D.; Lampert, H.C.; Drumm, D.; Roher, A.E. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1–42 levels. Biochem. Biophys. Res. Commun. 1998, 252, 711–715. [Google Scholar] [CrossRef]
- Sáiz-Vazquez, O.; Puente-Martínez, A.; Ubillos-Landa, S.; Pacheco-Bonrostro, J.; Santabárbara, J. Cholesterol and Alzheimer’s Disease Risk: A Meta-Meta-Analysis. Brain Sci. 2020, 10, 386. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Bodovitz, S.; Klein, W.L. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 1996, 271, 4436–4440. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef]
- Ghribi, O.; Larsen, B.; Schrag, M.; Herman, M.M. High cholesterol content in neurons increases BACE, beta-amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp. Neurol. 2006, 200, 460–467. [Google Scholar] [CrossRef]
- Cordy, J.M.; Hussain, I.; Dingwall, C.; Hooper, N.M.; Turner, A.J. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2003, 100, 11735–11740. [Google Scholar] [CrossRef]
- Song, Y.; Kenworthy, A.K.; Sanders, C.R. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Sci. 2014, 23, 1–22. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef]
- Song, Y.; Hustedt, E.J.; Brandon, S.; Sanders, C.R. Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 2013, 52, 5051–5064. [Google Scholar] [CrossRef]
- Langness, V.F.; van der Kant, R.; Das, U.; Wang, L.; Chaves, R.D.S.; Goldstein, L.S.B. Cholesterol-lowering drugs reduce APP processing to Aβ by inducing APP dimerization. Mol. Biol. Cell 2021, 32, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, L.; Foster, L.; Straub, J.E.; Thirumalai, D. Impact of membrane lipid composition on the structure and stability of the transmembrane domain of amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2016, 113, E5281–E5287. [Google Scholar] [CrossRef]
- Li, C.-D.; Junaid, M.; Shan, X.; Wang, Y.; Wang, X.; Khan, A.; Wei, D.-Q. Effect of Cholesterol on C99 Dimerization: Revealed by Molecular Dynamics Simulations. Front. Mol. Biosci. 2022, 9, 872385. [Google Scholar] [CrossRef]
- Sun, F.; Chen, L.; Wei, P.; Chai, M.; Ding, X.; Xu, L.; Luo, S.-Z. Dimerization and Structural Stability of Amyloid Precursor Proteins Affected by the Membrane Microenvironments. J. Chem. Inf. Modeling 2017, 57, 1375–1387. [Google Scholar] [CrossRef]
- Miyashita, N.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J. Am. Chem. Soc. 2009, 131, 3438–3439. [Google Scholar] [CrossRef]
- Gorman, P.M.; Kim, S.; Guo, M.; Melnyk, R.A.; McLaurin, J.; Fraser, P.E.; Bowie, J.U.; Chakrabartty, A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer’s disease mutants. BMC Neurosci. 2008, 9, 17. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, T.H.; Harikumar, K.G.; Miller, L.J.; Melcher, K.; Xu, H.E. Dimerization of the transmembrane domain of amyloid precursor protein is determined by residues around the γ-secretase cleavage sites. J. Biol. Chem. 2017, 292, 15826–15837. [Google Scholar] [CrossRef]
- Muller-Schiffmann, A.; Herring, A.; Abdel-Hafiz, L.; Chepkova, A.N.; Schable, S.; Wedel, D.; Horn, A.H.; Sticht, H.; de Souza Silva, M.A.; Gottmann, K.; et al. Amyloid-beta dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain 2016, 139, 509–525. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid beta Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. 2014, 6, 28. [Google Scholar] [CrossRef]
- Domingo, G.; Benussi, L.; Saraceno, C.; Bertuzzi, M.; Nicsanu, R.; Longobardi, A.; Bellini, S.; Cagnotto, A.; Salmona, M.; Binetti, G.; et al. N-Terminally Truncated and Pyroglutamate-Modified Abeta Forms Are Measurable in Human Cerebrospinal Fluid and Are Potential Markers of Disease Progression in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 708119. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wullner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef]
- Kumar, S.; Wirths, O.; Stuber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brustle, O.; et al. Phosphorylation of the amyloid beta-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef]
- Nirmalraj, P.N.; List, J.; Battacharya, S.; Howe, G.; Xu, L.; Thompson, D.; Mayer, M. Complete aggregation pathway of amyloid beta (1–40) and (1–42) resolved on an atomically clean interface. Sci. Adv. 2020, 6, eaaz6014. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef]
- Vadukul, D.M.; Vrancx, C.; Burguet, P.; Contino, S.; Suelves, N.; Serpell, L.C.; Quinton, L.; Kienlen-Campard, P. An evaluation of the self-assembly enhancing properties of cell-derived hexameric amyloid-beta. Sci. Rep. 2021, 11, 11570. [Google Scholar] [CrossRef]
- Sato, T.; Kienlen-Campard, P.; Ahmed, M.; Liu, W.; Li, H.; Elliott, J.I.; Aimoto, S.; Constantinescu, S.N.; Octave, J.N.; Smith, S.O. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry 2006, 45, 5503–5516. [Google Scholar] [CrossRef]
- Misra, P.; Kodali, R.; Chemuru, S.; Kar, K.; Wetzel, R. Rapid alpha-oligomer formation mediated by the A beta C terminus initiates an amyloid assembly pathway. Nat. Commun. 2016, 7, 12419. [Google Scholar] [CrossRef] [PubMed]
- Beel, A.J.; Sakakura, M.; Barrett, P.J.; Sanders, C.R. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 2010, 1801, 975–982. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Synergistic interactions between repeats in tau protein and Abeta amyloids may be responsible for accelerated aggregation via polymorphic states. Biochemistry 2011, 50, 5172–5181. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Umeda, T.; Tomiyama, T.; Kitajima, E.; Idomoto, T.; Nomura, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci. 2012, 91, 1169–1176. [Google Scholar] [CrossRef]
- Lazar, A.N.; Bich, C.; Panchal, M.; Desbenoit, N.; Petit, V.W.; Touboul, D.; Dauphinot, L.; Marquer, C.; Laprévote, O.; Brunelle, A.; et al. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathol. 2013, 125, 133–144. [Google Scholar] [CrossRef]
- Gille, B.; Galuska, C.E.; Fuchs, B.; Peleg, S. Recent Advances in Studying Age-Associated Lipids Alterations and Dietary Interventions in Mammals. Front. Aging 2021, 2, 773795. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulos, N.; Suelves, N.; Perrin, F.; Vadukul, D.M.; Vrancx, C.; Constantinescu, S.N.; Kienlen-Campard, P. Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates. Biomedicines 2022, 10, 2753. https://doi.org/10.3390/biomedicines10112753
Papadopoulos N, Suelves N, Perrin F, Vadukul DM, Vrancx C, Constantinescu SN, Kienlen-Campard P. Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates. Biomedicines. 2022; 10(11):2753. https://doi.org/10.3390/biomedicines10112753
Chicago/Turabian StylePapadopoulos, Nicolas, Nuria Suelves, Florian Perrin, Devkee M. Vadukul, Céline Vrancx, Stefan N. Constantinescu, and Pascal Kienlen-Campard. 2022. "Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates" Biomedicines 10, no. 11: 2753. https://doi.org/10.3390/biomedicines10112753
APA StylePapadopoulos, N., Suelves, N., Perrin, F., Vadukul, D. M., Vrancx, C., Constantinescu, S. N., & Kienlen-Campard, P. (2022). Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates. Biomedicines, 10(11), 2753. https://doi.org/10.3390/biomedicines10112753