
Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models


Abstract
:1. Stroke
2. Ischemic Stroke
2.1. Risk Factors
2.2. Diagnosis
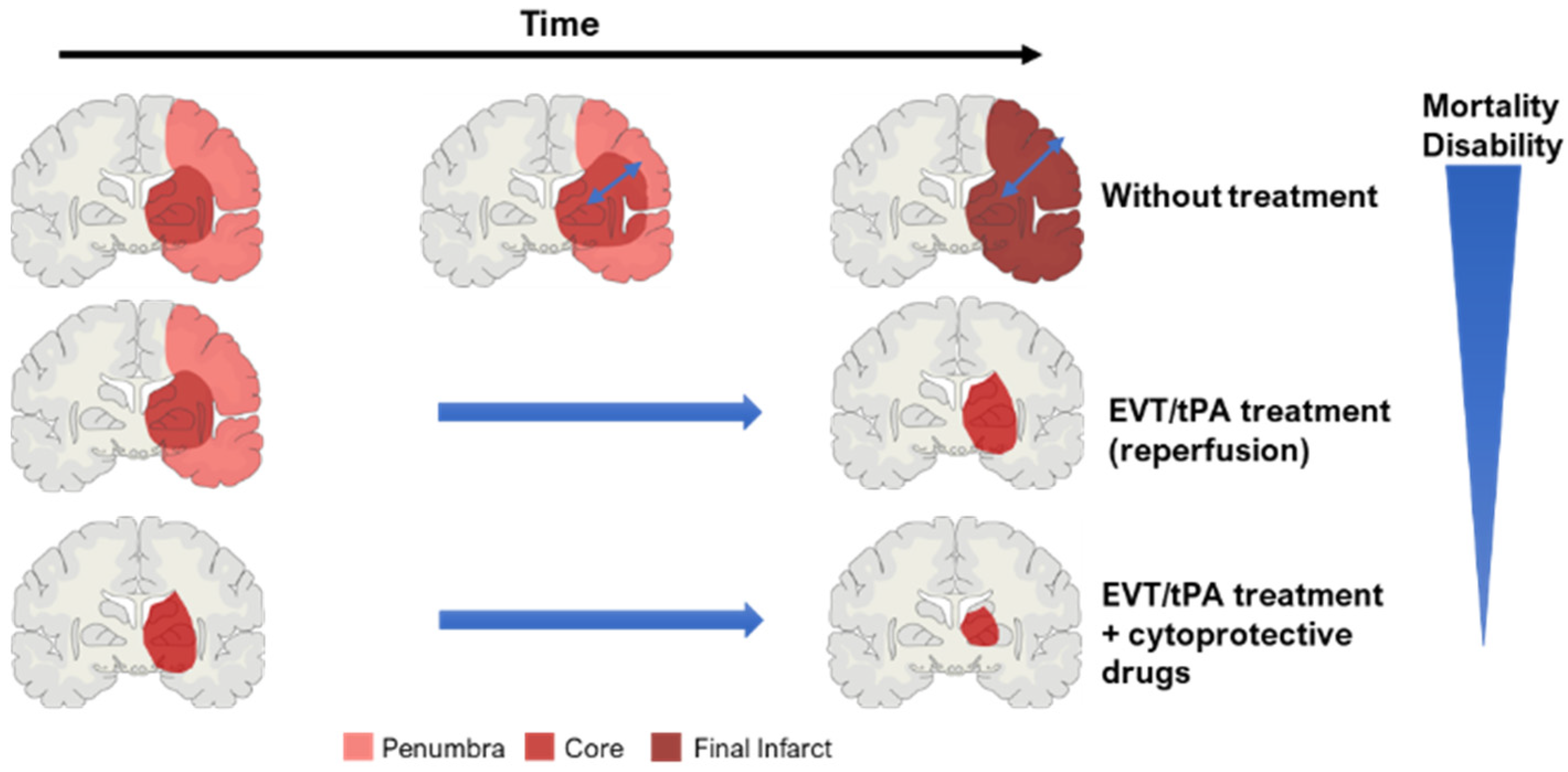
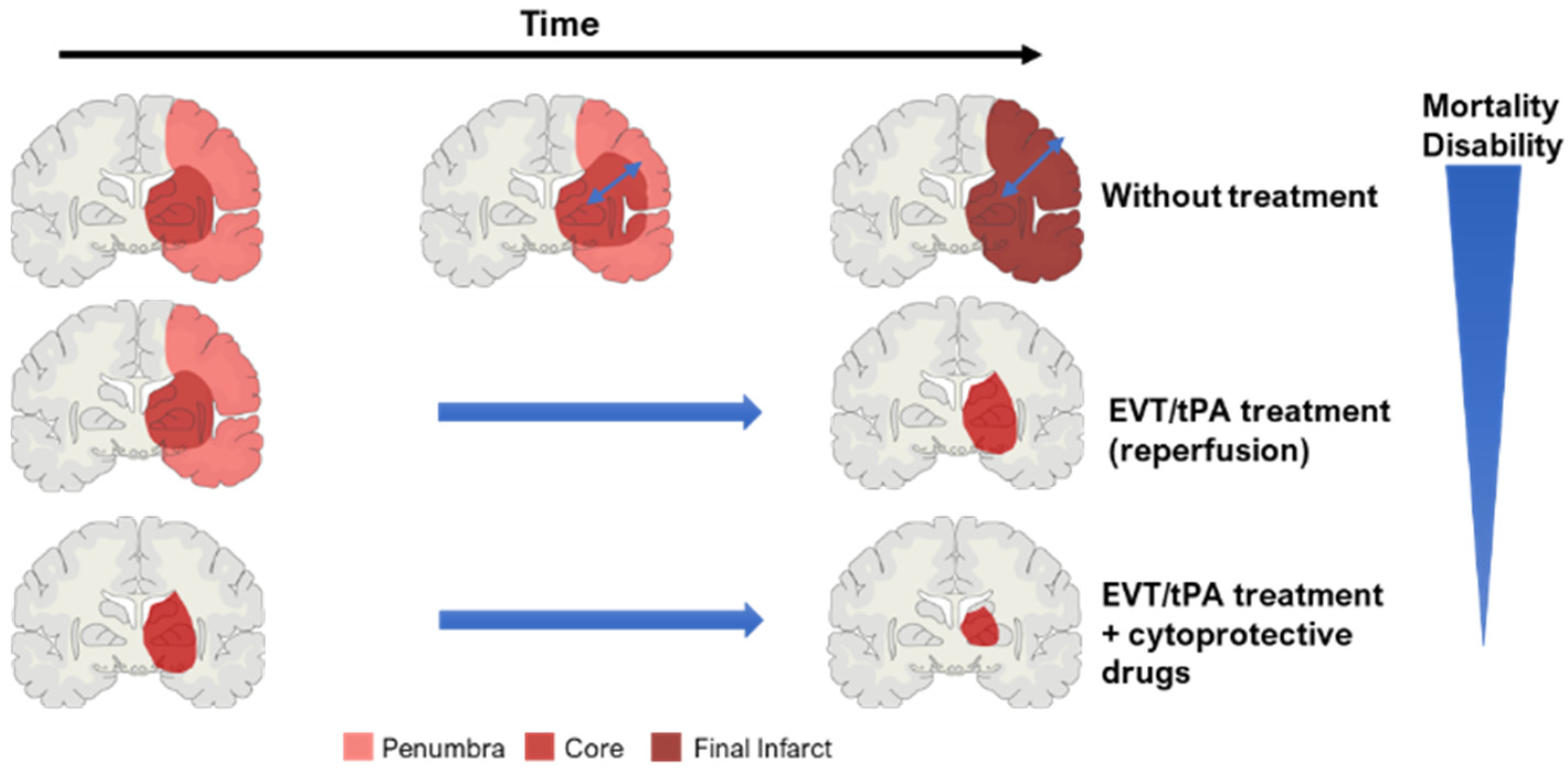
2.3. Key Concept: Core and Penumbra
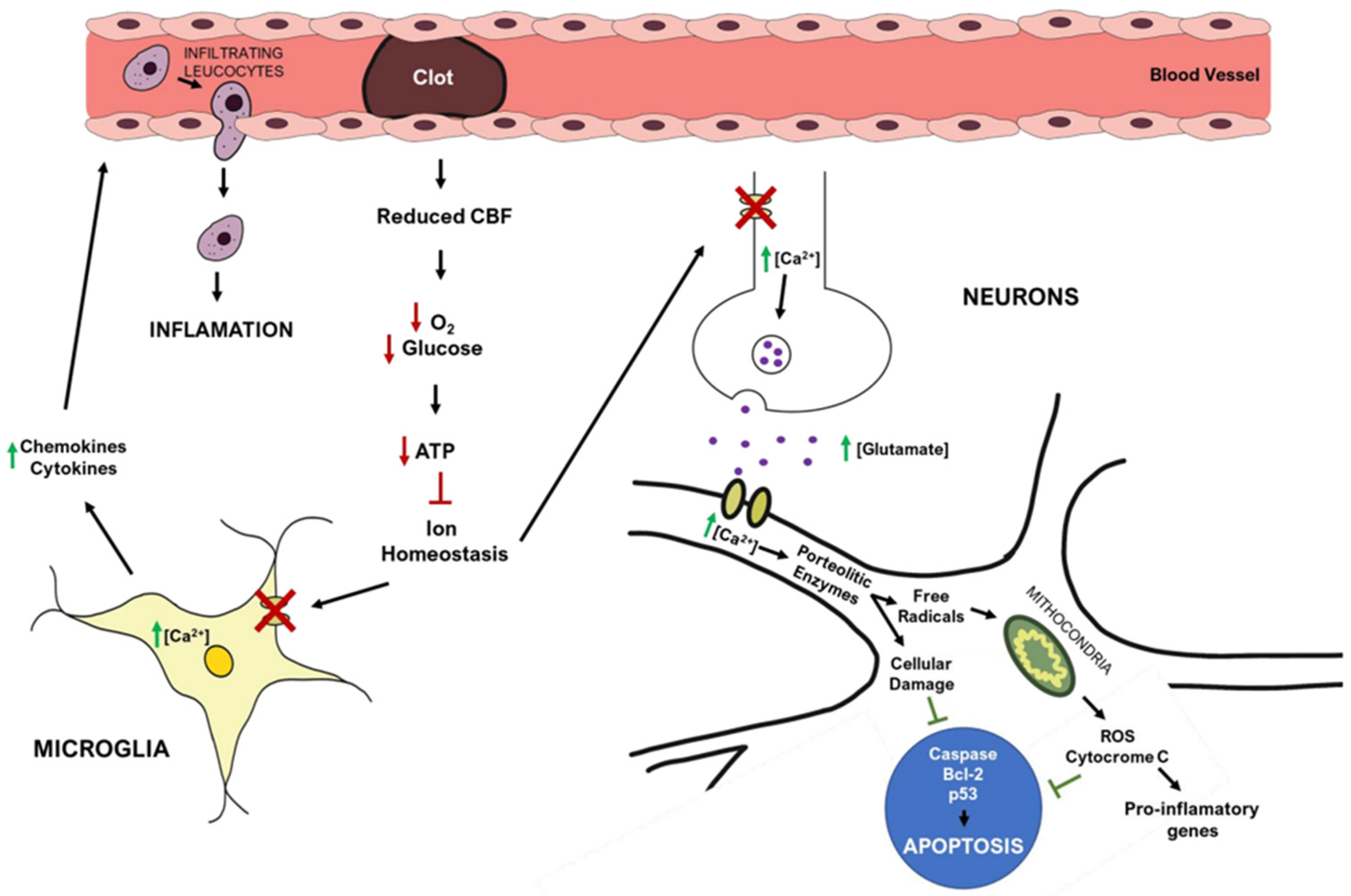
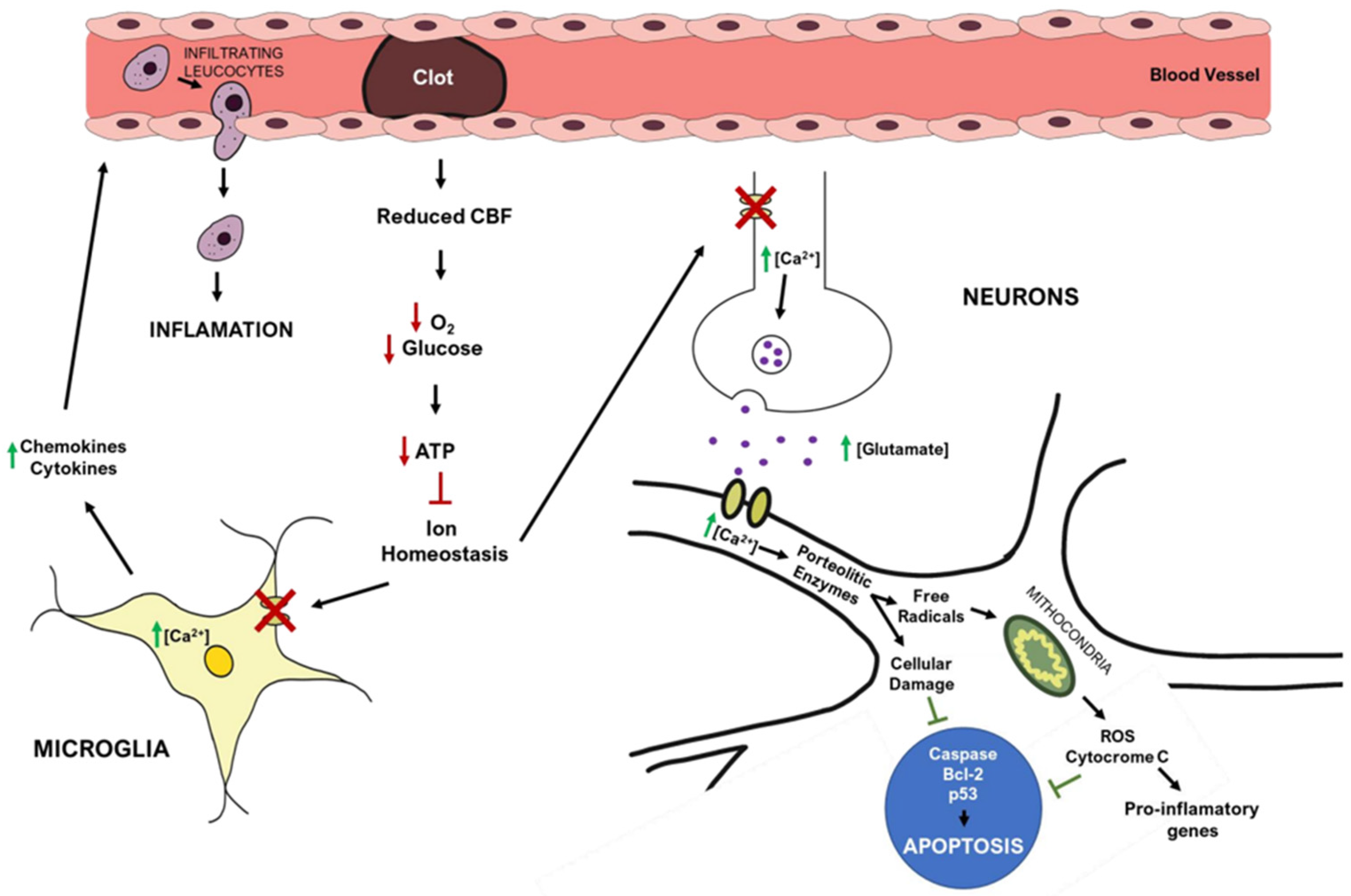
2.4. Ischemic Stroke Pathophysiology
2.5. Treatments
2.5.1. Thrombolysis
2.5.2. Mechanical Thrombectomy
2.5.3. Neuroprotective Drugs
2.6. Models for Acute Ischemic Stroke
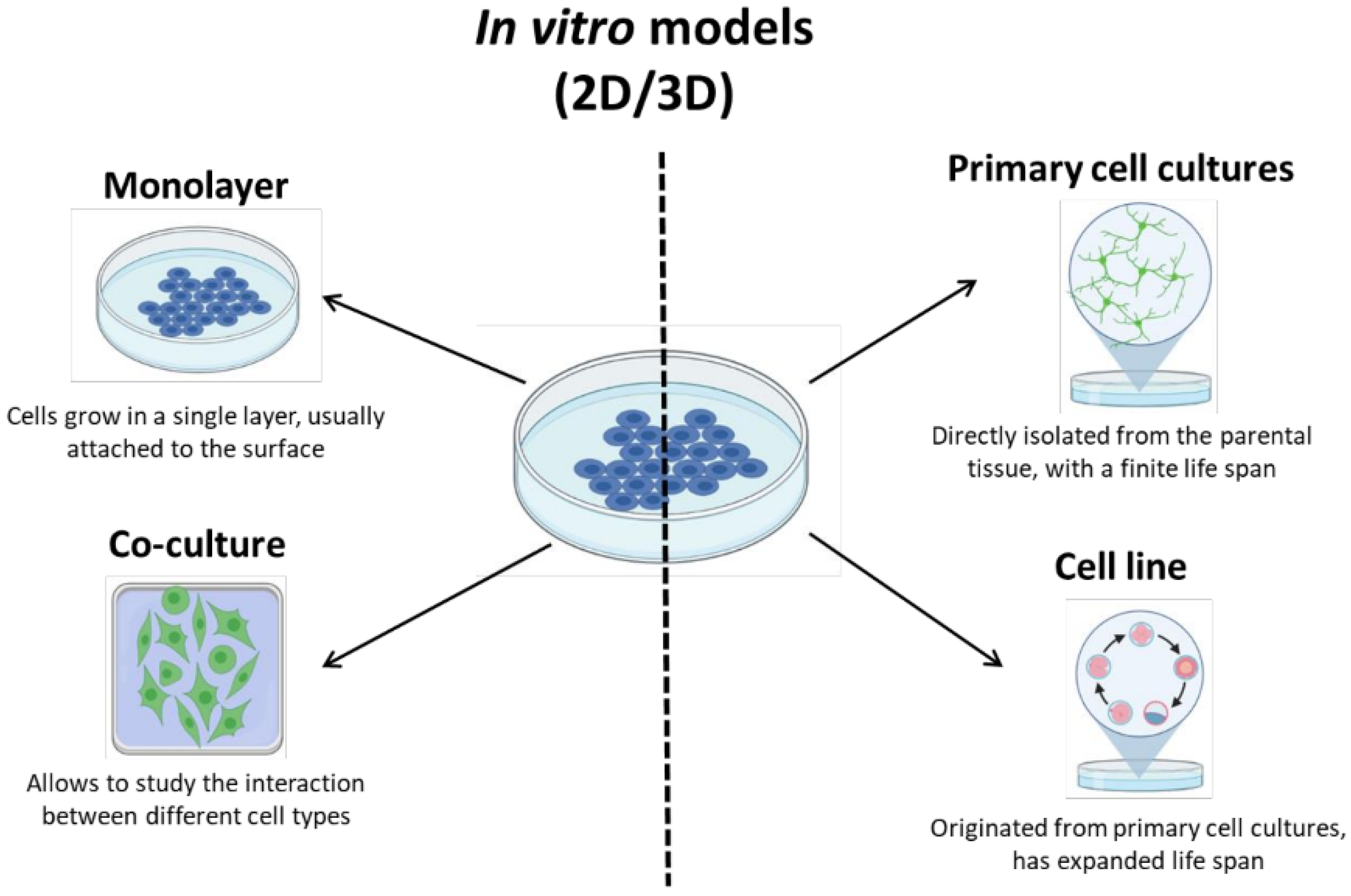
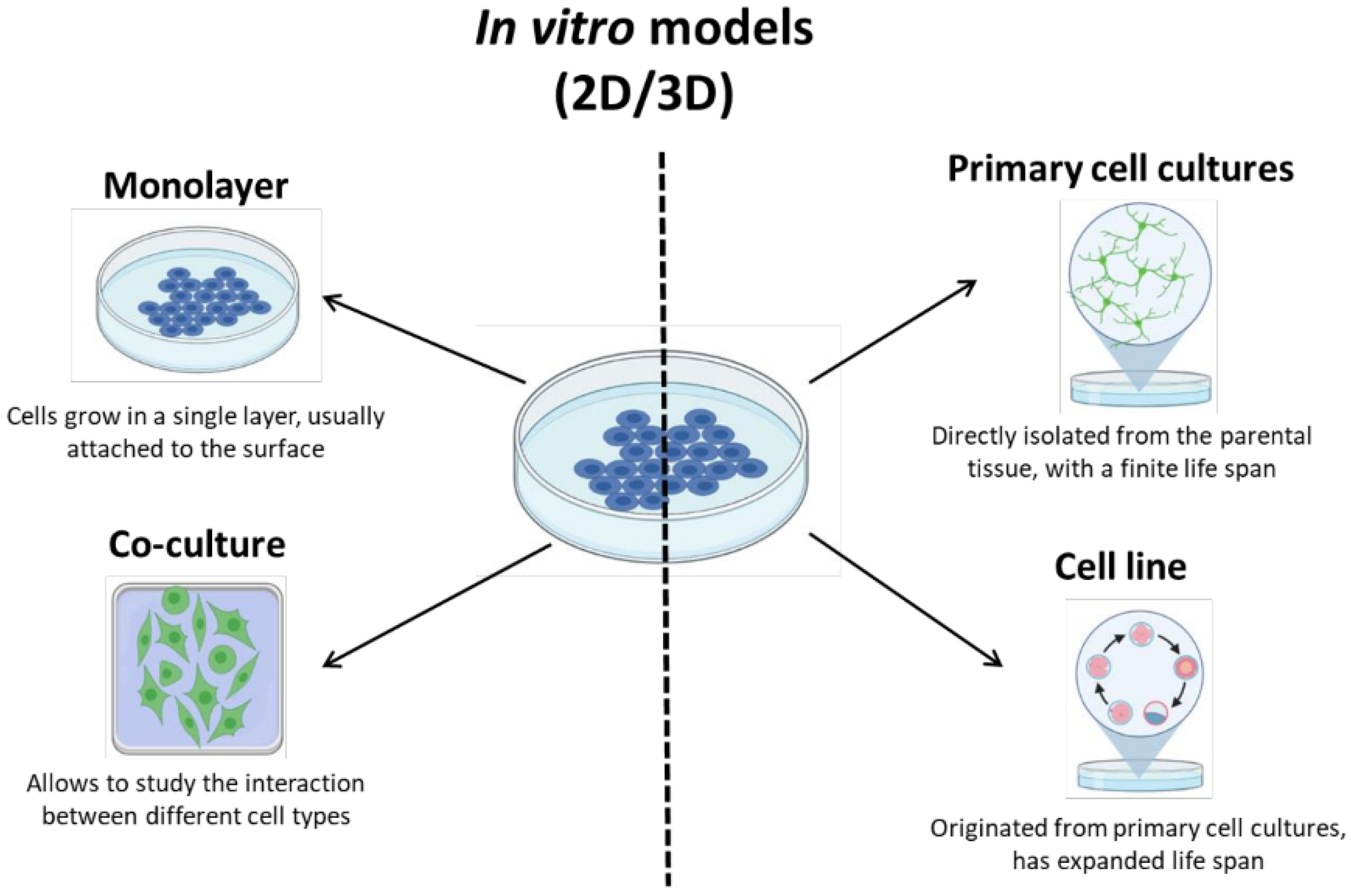
2.6.1. In Vitro Models for AIS
2.6.2. In Vivo Models for Acute Ischemic Stroke
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rtf, C. A Systematic Approach to the Definition of Stroke. Austin J. Cerebrovasc. Dis. Stroke 2014, 1, 1024. [Google Scholar]
- Coupland, A.P.; Thapar, A.; Qureshi, M.I.; Jenkins, H.; Davies, A.H. The Definition of Stroke. J. R. Soc. Med. 2017, 110, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Murphy, S.J.; Werring, D.J. Stroke: Causes and Clinical Features. Medicine 2020, 48, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.-z.; Zhang, S.-t.; Lei, P. Mechanisms of Neuronal Cell Death in Ischemic Stroke and Their Therapeutic Implications. In Medicinal Research Reviews; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, Regional, and National Burden of Stroke, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Wafa, H.A.; Wolfe, C.D.A.; Emmett, E.; Roth, G.A.; Johnson, C.O.; Wang, Y. Burden of Stroke in Europe: Thirty-Year Projections of Incidence, Prevalence, Deaths, and Disability-Adjusted Life Years. Stroke 2020, 51, 2418–2427. [Google Scholar] [CrossRef]
- Stevens, E.; Emmett, E.; Wang, Y.; McKevitt, C.; Wolfe, C.D.A. The Burden of Stroke in Europe: Report; Stroke Alliance for Europe: City Road, London, 2017. [Google Scholar]
- Kisling, L.A.; Das, J.M. Prevention Strategies; StatPearls Publ: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537222/ (accessed on 4 May 2022).
- Ntaios, G. Embolic Stroke of Undetermined Source: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 333–340. [Google Scholar] [CrossRef]
- Pierik, R.; Algra, A.; van Dijk, E.; Erasmus, M.E.; van Gelder, I.C.; Koudstaal, P.J.; Luijckx, G.R.; Nederkoorn, P.J.; van Oostenbrugge, R.J.; Ruigrok, Y.M.; et al. Distribution of Cardioembolic Stroke: A Cohort Study. Cere 2020, 49, 97–104. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic Stroke: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [Green Version]
- Gormley, G.J. Chemoprevention Strategies. J. Cell. Biochem. 1992, 50, 106. [Google Scholar] [CrossRef]
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of Ischemic and Hemorrhagic Stroke: Incidence, Prevalence, Mortality, and Risk Factors. Neurol. Clin. 2008, 26, 871–895. [Google Scholar] [CrossRef]
- National Institute of Health. Stroke|NHLBI, NIH; National Heart, Lung and Blood institute: Bethesda, MD, USA, 2008. [Google Scholar]
- Avan, A.; Digaleh, H.; Di Napoli, M.; Stranges, S.; Behrouz, R.; Shojaeianbabaei, G.; Amiri, A.; Tabrizi, R.; Mokhber, N.; Spence, J.D.; et al. Socioeconomic Status and Stroke Incidence, Prevalence, Mortality, and Worldwide Burden: An Ecological Analysis from the Global Burden of Disease Study 2017. BMC Med. 2019, 17, 191. [Google Scholar] [CrossRef] [Green Version]
- Howard, V.J.; Madsen, T.E.; Kleindorfer, D.O.; Judd, S.E.; Rhodes, J.D.; Soliman, E.Z.; Kissela, B.M.; Safford, M.M.; Moy, C.S.; McClure, L.A.; et al. Sex and Race Differences in the Association of Incident Ischemic Stroke with Risk Factors. JAMA Neurol. 2019, 76, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Floßmann, E.; Schulz, U.G.R.; Rothwell, P.M. Systematic Review of Methods and Results of Studies of the Genetic Epidemiology of Ischemic Stroke. Stroke 2004, 35, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Deb, P.; Sharma, S.; Hassan, K.M. Pathophysiologic Mechanisms of Acute Ischemic Stroke: An Overview with Emphasis on Therapeutic Significance beyond Thrombolysis. Pathophysiology 2010, 17, 197–218. [Google Scholar] [CrossRef]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Barthels, D.; Das, H. Current Advances in Ischemic Stroke Research and Therapies. Biochim. Biophys. Acta—Mol. Basis Dis. 2020, 1866, 165260. [Google Scholar] [CrossRef]
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart Disease and Stroke Statistics-2008 Update: A Report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008, 117, e25–e146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. In Circulation; Lippincott Williams & Wilkins: Hagerstown, MD, USA, 2020; pp. E139–E596. [Google Scholar] [CrossRef]
- WHO. WHO|Stroke, Cerebrovascular Accident|Health Topics; World Health Organization: Geneva, Switzerland, 2022.
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Randolph, S.A. Ischemic Stroke. Work. Health Saf. 2016, 64, 444. [Google Scholar] [CrossRef]
- Ojaghihaghighi, S.; Vahdati, S.S.; Mikaeilpour, A.; Ramouz, A. Comparison of Neurological Clinical Manifestation in Patients with Hemorrhagic and Ischemic Stroke. World J. Emerg. Med. 2017, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Kuhrij, L.S.; Marang-Van De Mheen, P.J.; Van Den Berg-Vos, R.M.; De Leeuw, F.E.; Nederkoorn, P.J.; Lingsma, H.F.; De Borst, G.J.; Van Norden, A.G.W.; Eysink Smeets, M.M.; Aerden, L.A.M.; et al. Determinants of Extended Door-to-Needle Time in Acute Ischemic Stroke and Its Influence on in-Hospital Mortality: Results of a Nationwide Dutch Clinical Audit. BMC Neurol. 2019, 19, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, D.G. Diagnosis of Ischemic Stroke. Am. J. Med. 2009, 122 (Suppl. S2), S14–S20. [Google Scholar] [CrossRef]
- Phipps, M.S.; Cronin, C.A. Management of Acute Ischemic Stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef]
- Puig, J.; Shankar, J.; Liebeskind, D.; Terceño, M.; Nael, K.; Demchuk, A.M.; Menon, B.; Dowlatshahi, D.; Leiva-Salinas, C.; Wintermark, M.; et al. From “Time Is Brain” to “Imaging Is Brain”: A Paradigm Shift in the Management of Acute Ischemic Stroke. J. Neuroimaging 2020, 30, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Calderon, V.J.; Kasturiarachi, B.M.; Lin, E.; Bansal, V.; Zaidat, O.O. Review of the Mobile Stroke Unit Experience Worldwide. Interv. Neurol. 2018, 7, 347–358. [Google Scholar] [CrossRef]
- Helwig, S.A.; Ragoschke-Schumm, A.; Schwindling, L.; Kettner, M.; Roumia, S.; Kulikovski, J.; Keller, I.; Manitz, M.; Martens, D.; Grün, D.; et al. Prehospital Stroke Management Optimized by Use of Clinical Scoring vs Mobile Stroke Unit for Triage of Patients with Stroke: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Kiewert, C.; Mdzinarishvili, A.; Hartmann, J.; Bickel, U.; Klein, J. Metabolic and Transmitter Changes in Core and Penumbra after Middle Cerebral Artery Occlusion in Mice. Brain Res. 2010, 1312, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, E.; Huțanu, A.; Chiriac, L.; Dobreanu, M.; Orădan, A.; Nagy, E.E. Ischemic Damage and Early Inflammatory Infiltration Are Different in the Core and Penumbra Lesions of Rat Brain after Transient Focal Cerebral Ischemia. J. Neuroimmunol. 2018, 324, 35–42. [Google Scholar] [CrossRef]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A New Era for Stroke Therapy: Integrating Neurovascular Protection with Optimal Reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef]
- Uzdensky, A.B. Apoptosis Regulation in the Penumbra after Ischemic Stroke: Expression of pro- and Antiapoptotic Proteins. In Apoptosis; Springer: Berlin/Heidelberg, Germany, 2019; pp. 687–702. [Google Scholar] [CrossRef]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke. In Clinical Interventions in Aging; Dove Press: Macclesfield, UK, 2020; pp. 469–484. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, P.; Guo, Q.; Shang, K.; Yan, D.; Du, S.; Lu, Y. Pathophysiology and Biomarkers in Acute Ischemic Stroke—A Review. Trop. J. Pharm. Res. 2013, 12, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Dirnagl, U.; Kunz, A. Ischemic Stroke: Basic Pathophysiology and Clinical Implication. In Neuroscience in the 21st Century—From Basic to Clinical; Springer: New York, NY, USA, 2013; pp. 2543–2563, Chapter 97. [Google Scholar] [CrossRef]
- Singhal, A.B.; Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Ischemic Stroke: Basic Pathophysiology and Neuroprotective Strategies. Acute Ischemic Stroke—Imaging and Intervention; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–24, Chapter 1. [Google Scholar] [CrossRef]
- Nour, M.; Scalzo, F.; Liebeskind, D.S. Ischemia-Reperfusion Injury in Stroke. Interv. Neurol. 2013, 1, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Meretoja, A.; Donnan, G.A.; Davis, S.M. Twenty-Year History of the Evolution of Stroke Thrombolysis with Intravenous Alteplase to Reduce Long-Term Disability. Stroke 2015, 46, 2341–2346. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.A.; Saver, J.L. Extending Reperfusion Therapy for Acute Ischemic Stroke: Emerging Pharmacological, Mechanical, and Imaging Strategies. Stroke 2005, 36, 2311–2320. [Google Scholar] [CrossRef]
- Bhaskar, S.; Stanwell, P.; Cordato, D.; Attia, J.; Levi, C. Reperfusion Therapy in Acute Ischemic Stroke: Dawn of a New Era? BMC Neurol. 2018, 18, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Daniel, M.; Goel, I.; Sakai, Y.; Teramura, Y. Current Status of Ischemic Stroke Treatment: From Thrombolysis to Potential Regenerative Medicine. Regen. Ther. 2021, 18, 408–417. [Google Scholar] [CrossRef]
- Herpich, F.; Rincon, F. Management of Acute Ischemic Stroke. Crit. Care Med. 2020, 48, 1654–1663. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Mitchell, P.J.; Churilov, L.; Yassi, N.; Kleinig, T.J.; Dowling, R.J.; Yan, B.; Bush, S.J.; Dewey, H.M.; Thijs, V.; et al. Tenecteplase versus Alteplase before Thrombectomy for Ischemic Stroke. N. Engl. J. Med. 2018, 378, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Berge, E.; Whiteley, W.; Audebert, H.; De Marchis, G.M.; Fonseca, A.C.; Padiglioni, C.; de la Ossa, N.P.; Strbian, D.; Tsivgoulis, G.; Turc, G. European Stroke Organisation (ESO) Guidelines on Intravenous Thrombolysis for Acute Ischaemic Stroke. Eur. Stroke J. 2021, 6, I. [Google Scholar] [CrossRef]
- Seners, P.; Caroff, J.; Chausson, N.; Turc, G.; Denier, C.; Piotin, M.; Aghasaryan, M.; Alecu, C.; Chassin, O.; Lapergue, B.; et al. Recanalization before Thrombectomy in Tenecteplase vs. Alteplase-Treated Drip-and-Ship Patients. J. Stroke 2019, 21, 105–107. [Google Scholar] [CrossRef]
- Papanagiotou, P.; Ntaios, G. Endovascular Thrombectomy in Acute Ischemic Stroke. Circ. Cardiovasc. Interv. 2018, 11, e005362. [Google Scholar] [CrossRef]
- Blanc, R.; Escalard, S.; Baharvadhat, H.; Desilles, J.P.; Boisseau, W.; Fahed, R.; Redjem, H.; Ciccio, G.; Smajda, S.; Maier, B.; et al. Recent Advances in Devices for Mechanical Thrombectomy. Expert Rev. Med. Devices 2020, 17, 697–706. [Google Scholar] [CrossRef]
- Maurice, A.; Ferré, J.C.; Ronzière, T.; Devys, J.M.; Subileau, A.; Laffon, M.; Laviolle, B.; Beloeil, H. GASS Trial Study Protocol: A Multicentre, Single-Blind, Randomised Clinical Trial Comparing General Anaesthesia and Sedation during Intra-Arterial Treatment for Stroke. BMJ Open 2019, 9, e024249. [Google Scholar] [CrossRef] [PubMed]
- Tsivgoulis, G.; Katsanos, A.H. Important Advances in Stroke Research in 2020. Lancet Neurol. 2021, 20, 2–3. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, Y.; Zhang, L.; Zhang, Y.; Treurniet, K.M.; Chen, W.; Peng, Y.; Han, H.; Wang, J.; Wang, S.; et al. Endovascular Thrombectomy with or without Intravenous Alteplase in Acute Stroke. N. Engl. J. Med. 2020, 382, 1981–1993. [Google Scholar] [CrossRef]
- Katsanos, A.H.; Malhotra, K.; Goyal, N.; Arthur, A.; Schellinger, P.D.; Köhrmann, M.; Krogias, C.; Turc, G.; Magoufis, G.; Leys, D.; et al. Intravenous Thrombolysis Prior to Mechanical Thrombectomy in Large Vessel Occlusions. Ann. Neurol. 2019, 86, 395–406. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Tsivgoulis, G. Large Vessel Occlusion Strokes after the DIRECT-MT and SKIP Trials: Is the Alteplase Syringe Half Empty or Half Full? Stroke 2020, 51, 3182–3186. [Google Scholar] [CrossRef]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. Neuroprotectants in Stroke. Pharmacotherapy 2008, 9, 887–900. [Google Scholar] [CrossRef]
- DeGraba, T.J.; Pettigrew, L.C. Why Do Neuroprotective Drugs Work in Animals but Not Humans? Neurol. Clin. 2000, 18, 475–493. [Google Scholar] [CrossRef]
- Dirnagl, U.; Priller, J. Focal Cerebral Ischemia: The Multifaceted Role of Glial Cells. In Neuroglia; Oxford Academic: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Nedergaard, M.; Dirnagl, U. Role of Glial Cells in Cerebral Ischemia. Glia 2005, 50, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A Critical Role in Ischemic Stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tang, H.; Li, H.; Zhao, R.; Huang, Q.; Liu, J. Recent Advances in the Development of Neuroprotective Agents and Therapeutic Targets in the Treatment of Cerebral Ischemia. Eur. J. Med. Chem. 2019, 162, 132–146. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Zhang, J.H. The High Cost of Stroke and Stroke Cytoprotection Research. Transl. Stroke Res. 2016, 8, 307–317. [Google Scholar] [CrossRef]
- Fisher, M.; Savitz, S.I. Pharmacological Brain Cytoprotection in Acute Ischaemic Stroke—Renewed Hope in the Reperfusion Era. Nat. Rev. Neurol. 2022, 18, 193–202. [Google Scholar] [CrossRef]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for Ischemic Stroke: Two Decades of Success and Failure. NeuroRx 2004, 1, 36. [Google Scholar] [CrossRef]
- Das, J.M.; Zito, P.M. Nimodipine. In xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 1–7. [Google Scholar] [CrossRef]
- Horn, J.; De Haan, R.J.; Vermeulen, M.; Limburg, M. Very Early Nimodipine Use in Stroke (VENUS). Stroke 2001, 32, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Magnesium and Verapamil after Recanalization in Ischemia of the Cerebrum: A Clinical and Translational Study (MAVARIC; NCT02912663). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT02912663&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital Use of Magnesium Sulfate as Neuroprotection in Acute Stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Cigel, A.; Sayin, O.; Gurgen, S.G.; Sonmez, A. Long Term Neuroprotective Effects of Acute Single Dose MK-801treatment against Traumatic Brain Injury in Immature Rats. Neuropeptides 2021, 88, 102161. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific Coupling of NMDA Receptor Activation to Nitric Oxide Neurotoxicity by PSD-95 Protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Ugalde-Triviño, L.; Díaz-Guerra, M. PSD-95: An Effective Target for Stroke Therapy Using Neuroprotective Peptides. Int. J. Mol. Sci. 2021, 22, 12585. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor-PSD-95 Protein Interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Fisher, M.; Israel, B.; Xiong, Y.; Wakhloo, A.K. Advances in Acute Ischemic Stroke Therapy. Circ. Res. 2022, 130, 1230–1251. [Google Scholar] [CrossRef]
- Efficacy and Safety of Nerinetide in Participants with Acute Ischemic Stroke Undergoing Endovascular Thrombectomy Excluding Thrombolysis.(ESCAPE-NEXT; NCT04462536). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04462536&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Bennion, D.M.; Isenberg, J.D.; Harmel, A.T.; DeMars, K.; Dang, A.N.; Jones, C.H.; Pignataro, M.E.; Graham, J.T.; Steckelings, U.M.; Alexander, J.C.; et al. Post-Stroke Angiotensin II Type 2 Receptor Activation Provides Long-Term Neuroprotection in Aged Rats. PLoS ONE 2017, 12, e0180738. [Google Scholar] [CrossRef] [Green Version]
- Eklund, P.C.; Långvik, O.K.; Wärnå, J.P.; Salmi, T.O.; Willför, S.M.; Sjöholm, R.E. Chemical Studies on Antioxidant Mechanisms and Free Radical Scavenging Properties of Lignans. Org. Biomol. Chem. 2005, 3, 3336–3347. [Google Scholar] [CrossRef]
- Aliena-Valero, A.; Rius-Pérez, S.; Baixauli-Martín, J.; Torregrosa, G.; Chamorro, Á.; Pérez, S.; Salom, J.B. Uric Acid Neuroprotection Associated to IL-6/STAT3 Signaling Pathway Activation in Rat Ischemic Stroke. Mol. Neurobiol. 2020, 58, 408–423. [Google Scholar] [CrossRef]
- Chamorro, Á.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Martí-Fábregas, J.; Gállego, J.; Krupinski, J.; Gomis, M.; Cánovas, D.; et al. Safety and Efficacy of Uric Acid in Patients with Acute Stroke (URICO-ICTUS): A Randomised, Double-Blind Phase 2b/3 Trial. Lancet Neurol. 2014, 13, 453–460. [Google Scholar] [CrossRef]
- Chamorro, Á.; Lo, E.H.; Renú, A.; Van Leyden, K.; Lyden, P.D. The Future of Neuroprotection in Stroke. J. Neurol. Neurosurg. Psychiatry 2021, 92, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of Butylphthalide for Acute Ischemic Stroke Patients Receiving Intravenous Thrombolysis or Endovascular Treatment. (BAST; NCT03539445). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT03539445&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in Acute Ischemic Stroke. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Otomo, E.; Tohgi, H.; Kogure, K.; Hirai, S.; Takakura, K.; Terashi, A.; Gotoh, F.; Maruyama, S.; Tazaki, Y.; Shinohara, Y.; et al. Effect of a Novel Free Radical Scavenger, Edaravone (MCI-186), on Acute Brain Infarction. Randomized, Placebo-Controlled, Double-Blind Study at Multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar] [CrossRef]
- Chao, J.; Chao, L. Experimental Therapy with Tissue Kallikrein against Cerebral Ischemia. Front. Biosci. 2006, 11, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Lyu, Y.; Yang, X.L.; Chen, X.; Zhong, P.; Wu, D. Therapeutic Values of Human Urinary Kallidinogenase on Cerebrovascular Diseases. Front. Neurol. 2018, 9, 403. [Google Scholar] [CrossRef] [Green Version]
- Evaluation of HUK in Acute Stroke Patients: MRS and CTP (NCT0331909). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Evaluation+of+HUK+in+Acute+Stroke+Patients%3A+MRS+and+CTP&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Mulder, I.A.; van Bavel, E.T.; de Vries, H.E.; Coutinho, J.M. Adjunctive Cytoprotective Therapies in Acute Ischemic Stroke: A Systematic Review. Fluids Barriers CNS 2021, 18, 46. [Google Scholar] [CrossRef]
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Talhada, D.; Gonçalves, I.; Santos, C.R.; Ruscher, K. Transthyretin Expression in the Postischemic Brain. PLoS ONE 2019, 14, e0221555. [Google Scholar] [CrossRef] [Green Version]
- Clinical Trial on Ischemic Stroke: Allogeneic adult Mesenchymal Bone Marrow Stem Cells—Clinical Trials Registry—ICH GCP. Available online: https://ichgcp.net/clinical-trials-registry/NCT01297413 (accessed on 8 September 2022).
- Allogeneic Mesenchymal Stem Cells for the Survivors of Ischemic Stroke Trial (ASSIST; NCT04590118). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04590118&cntry=&state=&city=&dist= (accessed on 8 September 2022).
- Levy, M.L.; Crawford, J.R.; Dib, N.; Verkh, L.; Tankovich, N.; Cramer, S.C. Phase I/II Study of Safety and Preliminary Efficacy of Intravenous Allogeneic Mesenchymal Stem Cells in Chronic Stroke. Stroke 2019, 50, 2835–2841. [Google Scholar] [CrossRef]
- Suda, S.; Nito, C.; Ihara, M.; Iguchi, Y.; Urabe, T.; Matsumaru, Y.; Sakai, N.; Kimura, K. Randomised Placebo-Controlled Multicentre Trial to Evaluate the Efficacy and Safety of JTR-161, Allogeneic Human Dental Pulp Stem Cells, in Patients with Acute Ischaemic StRoke (J-REPAIR). BMJ Open 2022, 12, e054269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Fu, Y.; Tian, D.; Sun, N.; Han, W.; Chang, G.; Dong, Y.; Xu, X.; Liu, Q.; Huang, D.; et al. Combination of the Immune Modulator Fingolimod with Alteplase in Acute Ischemic Stroke: A Pilot Trial. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of FTY720 for Acute Stroke (NCT02002390). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT02002390&cntry=&state=&city=&dist= (accessed on 8 September 2022).
- Combination of the Immune Modulator Dimethyl Fumarate with Alteplase in Acute Ischemic Stroke (NCT04890366). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04890366&cntry=&state=&city=&dist= (accessed on 8 September 2022).
- Liu, C.W.; Liao, K.H.; Wu, C.M.; Chen, H.Y.; Wang, E.Y.; Lai, T.W. Stroke Injury Induced by Distal Middle Cerebral Artery Occlusion Is Resistant to N-Methyl-d-Aspartate Receptor Antagonism in FVB/NJ Mice. Neuroreport 2021, 32, 1122–1127. [Google Scholar] [CrossRef]
- Jickling, G.C.; Sharp, F.R. Improving the Translation of Animal Ischemic Stroke Studies to Humans. Metab. Brain Dis. 2015, 30, 461–467. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The Arrive Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, 6–10. [Google Scholar] [CrossRef]
- Holloway, P.M.; Gavins, F.N.E. Modeling Ischemic Stroke in Vitro: Status Quo and Future Perspectives. In Stroke; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2016; pp. 561–569. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.; Asayama, Y. Animal-cell Culture Media: History, Characteristics, and Current Issues. Reprod. Med. Biol. 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human Primary Cells Versus Cell Lines: Differences and Advantages. Available online: https://promocell.com/in-the-lab/human-primary-cells-and-immortal-cell-lines/ (accessed on 12 July 2022).
- Brown, R.C.; Davis, T.P. Hypoxia/Aglycemia Alters Expression of Occludin and Actin in Brain Endothelial Cells. Biochem. Biophys. Res. Commun. 2005, 327, 1114–1123. [Google Scholar] [CrossRef]
- Goldberg, M.P.; Choi, D.W. Combined Oxygen and Glucose Deprivation in Cortical Cell Culture: Calcium-Dependent and Calcium-Independent Mechanisms of Neuronal Injury. J. Neurosci. 1993, 13, 3510–3524. [Google Scholar] [CrossRef] [PubMed]
- Gwag, B.J.; Lobner, D.; Koh, J.Y.; Wie, M.B.; Choi, D.W. Blockade of Glutamate Receptors Unmasks Neuronal Apoptosis after Oxygen-Glucose Deprivation in Vitro. Neuroscience 1995, 68, 615–619. [Google Scholar] [CrossRef]
- Kalda, A.; Eriste, E.; Vassiljev, V.; Zharkovsky, A. Medium Transitory Oxygen-Glucose Deprivation Induced Both Apoptosis and Necrosis in Cerebellar Granule Cells. Neurosci. Lett. 1998, 240, 21–24. [Google Scholar] [CrossRef]
- Kurian, G.; Pemaih, B. Standardization of in Vitro Cell-Based Model for Renal Ischemia and Reperfusion Injury. Indian J. Pharm. Sci. 2014, 76, 348. [Google Scholar]
- Fennema, E.; Rivron, N.; Rouwkema, J.; van Blitterswijk, C.; De Boer, J. Spheroid Culture as a Tool for Creating 3D Complex Tissues. In Trends in Biotechnology; Elsevier Current Trends: Amsterdam, The Netherlands, 2013; pp. 108–115. [Google Scholar] [CrossRef]
- Dong, W.Q.; Schurr, A.; Reid, K.H.; Shields, C.B.; West, C.A. The Rat Hippocampal Slice Preparation as an in Vitro Model of Ischemia. Stroke 1988, 19, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Dingle, Y.T.L.; Boutin, M.E.; Chirila, A.M.; Livi, L.L.; Labriola, N.R.; Jakubek, L.M.; Morgan, J.R.; Darling, E.M.; Kauer, J.A.; Hoffman-Kim, D. Three-Dimensional Neural Spheroid Culture: An In Vitro Model for Cortical Studies. Tissue Eng.—Part C Methods 2015, 21, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Nürnberg, E.; Vitacolonna, M.; Klicks, J.; von Molitor, E.; Cesetti, T.; Keller, F.; Bruch, R.; Ertongur-Fauth, T.; Riedel, K.; Scholz, P.; et al. Routine Optical Clearing of 3D-Cell Cultures: Simplicity Forward. Front. Mol. Biosci. 2020, 7, 20. [Google Scholar] [CrossRef]
- Kronemberger, G.S.; Carneiro, F.A.; Rezende, D.F.; Baptista, L.S. Spheroids and Organoids as Humanized 3D Scaffold-Free Engineered Tissues for SARS-CoV-2 Viral Infection and Drug Screening. Artif. Organs 2021, 45, 548–558. [Google Scholar] [CrossRef]
- Sreenivasalu, G.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]
- Ko, E.; Poon, M.L.S.; Park, E.; Cho, Y.; Shin, J.H. Engineering 3D Cortical Spheroids for an in Vitro Ischemic Stroke Model. ACS Biomater. Sci. Eng. 2021, 7, 3845–3860. [Google Scholar] [CrossRef]
- Kessel, S.; Cribbes, S.; Bonasu, S.; Rice, W.; Qiu, J.; Chan, L.L.Y. Real-Time Viability and Apoptosis Kinetic Detection Method of 3D Multicellular Tumor Spheroids Using the Celigo Image Cytometer. Cytom. Part A 2017, 91, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as in Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Nzou, G.; Wicks, R.T.; Wicks, E.E.; Seale, S.A.; Sane, C.H.; Chen, A.; Murphy, S.V.; Jackson, J.D.; Atala, A.J. Human Cortex Spheroid with a Functional Blood Brain Barrier for High-Throughput Neurotoxicity Screening and Disease Modeling. Sci. Rep. 2018, 8, 7413. [Google Scholar] [CrossRef] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In Vitro Models of the Blood-Brain Barrier: An Overview of Commonly Used Brain Endothelial Cell Culture Models and Guidelines for Their Use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Albanese, V.; Tommasino, C.; Spadaro, A.; Tomasello, F. A Transbasisphenoidal Approach for Selective Occlusion of the Middle Cerebral Artery in Rats. Experientia 1980, 36, 1302–1304. [Google Scholar] [CrossRef]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal Cerebral Ischaemia in the Rat: I. Description of Technique and Early Neuropathological Consequences Following Middle Cerebral Artery Occlusion. J. Cereb. Blood Flow Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Astrup, J.; Siesjo, B.k.; Symon, L. Thresholds in Cerebral Ischemia—The Ischemic Penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Endres, M. Found in Translation: Preclinical Stroke Research Predicts Human Pathophysiology, Clinical Phenotypes, and Therapeutic Outcomes. Stroke 2014, 45, 1510–1518. [Google Scholar] [CrossRef] [Green Version]
- Haley, M.J.; Lawrence, C.B. Obesity and Stroke: Can We Translate from Rodents to Patients? J. Cereb. Blood Flow Metab. 2016, 36, 2007–2021. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal Models of Ischemic Stroke and Their Application in Clinical Research. Drug Des. Devel. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [Green Version]
- Howells, D.W.; Porritt, M.J.; Rewell, S.S.J.; O’Collins, V.; Sena, E.S.; Van Der Worp, H.B.; Traystman, R.J.; MacLeod, M.R. Different Strokes for Different Folks: The Rich Diversity of Animal Models of Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1412–1431. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhen, G.; Meloni, B.P.; Campbell, K. Rodent stroke model guidelines for preclinical stroke trials (1st Edition). J. Exp. Stroke Transl. Med. 2009, 2, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Forsting, M.; Reith, W.; Scha, W.; Heiland, S.; Kummer, R. Von. Decompressive Craniectomy for Cerebral Infarction. Am. Heart Assoc. 1995, 26, 259–264. [Google Scholar]
- Engelhorn, T.; Doerfler, A.; Kastrup, A.; Beaulieu, C.; De Crespigny, A.; Forsting, M.; Moseley, M.E. Decompressive Craniectomy, Reperfusion, or a Combination for Early Treatment of Acute “Malignant” Cerebral Hemispheric Stroke in Rats? Stroke 1999, 30, 1456–1463. [Google Scholar] [CrossRef]
- Barber, P.A.; Hoyte, L.; Colbourne, F.; Buchan, A.M. Temperature-Regulated Model of Focal Ischemia in the Mouse: A Study with Histopathological and Behavioral Outcomes. Stroke 2004, 35, 1720–1725. [Google Scholar] [CrossRef] [Green Version]
- Kassem-Moussa, H.; Graffagnino, C. Nonocclusion and Spontaneous Recanalization Rates in Acute Ischemic Stroke: A Review of Cerebral Angiography Studies. Arch. Neurol. 2002, 59, 1870–1873. [Google Scholar] [CrossRef]
- Sutherland, B.A.; Neuhaus, A.A.; Couch, Y.; Balami, J.S.; Deluca, G.C.; Hadley, G.; Harris, S.L.; Grey, A.N.; Buchan, A.M. The Transient Intraluminal Filament Middle Cerebral Artery Occlusion Model as a Model of Endovascular Thrombectomy in Stroke. J. Cereb. Blood Flow Metab. 2016, 36, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Trotman-Lucas, M.; Gibson, C.L. A Review of Experimental Models of Focal Cerebral Ischemia Focusing on the Middle Cerebral Artery Occlusion Model. F1000Research 2021, 10, 242. [Google Scholar] [CrossRef]
- Goyal, M.; Menon, B.K.; Van Zwam, W.H.; Dippel, D.W.J.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.L.M.; Van Der Lugt, A.; De Miquel, M.A.; et al. Endovascular Thrombectomy after Large-Vessel Ischaemic Stroke: A Meta-Analysis of Individual Patient Data from Five Randomised Trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Dirnagl, U. Rodent Models of Stroke. In Neuromethods; Springer: Saskatoon, SK, Canada, 2016; Volume 120. [Google Scholar] [CrossRef]
- Yu, S.; Liu, X.; Shen, Y.; Xu, H.; Yang, Y.; Ding, F. Therapeutic Benefits of Combined Treatment with Tissue Plasminogen Activator and 2-(4-Methoxyphenyl)Ethyl-2-Acetamido-2-Deoxy-β-d-Pyranoside in an Animal Model of Ischemic Stroke. Neuroscience 2016, 327, 44–52. [Google Scholar] [CrossRef]
- Zhang, F.; Guo, R.M.; Yang, M.; Wen, X.H.; Shen, J. A Stable Focal Cerebral Ischemia Injury Model in Adult Mice: Assessment Using 7T MR Imaging. Am. J. Neuroradiol. 2012, 33, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Stehling, F.; Weber, R.; Özcelik, A.; Bröcker, M.; Volbracht, L.; Diener, H.C.; Busch, E. Acute Changes of Coagulation and Fibrinolysis Parameters after Experimental Thromboembolic Stroke and Thrombolytic Therapy. Neurosci. Lett. 2008, 441, 39–43. [Google Scholar] [CrossRef]
- MacRae, I. Preclinical Stroke Research—Advantages and Disadvantages of the Most Common Rodent Models of Focal Ischaemia. Br. J. Pharmacol. 2011, 164, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Horie, N.; Maag, A.-L.; Hamilton, S.A.; Shichinohe, H.; Bliss, T.M.; Steinberg, G.K. Mouse Model of Focal Cerebral Ischemia Using Endothelin-1. J. Neurosci. Methods 2008, 173, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Soleman, S.; Yip, P.; Leasure, J.L.; Moon, L. Sustained Sensorimotor Impairments after Endothelin-1 Induced Focal Cerebral Ischemia (Stroke) in Aged Rats. Exp. Neurol. 2010, 222, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Percie du Sert, N.; Alfieri, A.; Allan, S.M.; Carswell, H.V.O.; Deuchar, G.A.; Farr, T.D.; Flecknell, P.; Gallagher, L.; Gibson, C.L.; Haley, M.J.; et al. The IMPROVE Guidelines (Ischaemia Models: Procedural Refinements Of in Vivo Experiments). J. Cereb. Blood Flow Metab. 2017, 37, 3488–3517. [Google Scholar] [CrossRef] [Green Version]
- Rewell, S.S.J.; Churilov, L.; Sidon, T.K.; Aleksoska, E.; Cox, S.F.; Macleod, M.R.; Howells, D.W. Evolution of Ischemic Damage and Behavioural Deficit over 6 Months after MCAo in the Rat: Selecting the Optimal Outcomes and Statistical Power for Multi-Centre Preclinical Trials. PLoS ONE 2017, 12, e0171688. [Google Scholar] [CrossRef] [Green Version]
- Modo, M.; Stroemer, R.P.; Tang, E.; Veizovic, T.; Sowniski, P.; Hodges, H. Neurological Sequelae and Long-Term Behavioural Assessment of Rats with Transient Middle Cerebral Artery Occlusion. J. Neurosci. Methods 2000, 104, 99–109. [Google Scholar] [CrossRef]
- Balkaya, M.; Kröber, J.M.; Rex, A.; Endres, M. Assessing Post-Stroke Behavior in Mouse Models of Focal Ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Markgraf, C.G.; Green, E.J.; Watson, B.; Mccabe, P.M.; Schneiderman, N.; Dietrich, W.D.; Ginsberg, M.D. Recovery of Sensorimotor Function After Distal Middle Cerebral Artery Photothrombotic Occlusion in Rats. Stroke 1999, 25, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, J.; Li, Y.; Zhang, Z.G.; Chopp, M. Quantitative Measurement of Motor and Somatosensory Impairments after Mild (30 Min) and Severe (2 h) Transient Middle Cerebral Artery Occlusion in Rats. J. Neurol. Sci. 2000, 174, 141–146. [Google Scholar] [CrossRef]
- Freret, T.; Bouet, V.; Leconte, C.; Roussel, S.; Chazalviel, L.; Divoux, D.; Schumann-Bard, P.; Boulouard, M. Behavioral Deficits After Distal Focal Cerebral Ischemia in Mice: Usefulness of Adhesive Removal Test. Behav. Neurosci. 2009, 123, 224–230. [Google Scholar] [CrossRef]
- Hattori, K.; Lee, H.; Hurn, P.D.; Crain, B.J.; Traystman, R.J.; DeVries, A.C. Cognitive Deficits after Focal Cerebral Ischemia in Mice. Stroke 2000, 31, 1939–1944. [Google Scholar] [CrossRef]
- STAIR. Recommendations for Standards Regarding Preclinical Neuroprotective and Restorative Drug Development. Stroke 1999, 30, 2752–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, R.; Magalhães, A.; Sousa, M.; Melo, L.; Lobo, A.; Barros, P.; Gomes, J.R. Bridging the Transient Intraluminal Stroke Preclinical Model to Clinical Practice: From Improved Surgical Procedures to a Workflow of Functional Tests. Front. Neurol. 2022, 13, 846735. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Schulz, K.F.; Altman, D.G.; for the Group, C. The CONSORT Statement: Revised Recommendations for Improving the Quality of Reports of Parallel-Group Randomised Trials. Lancet 2001, 357, 1191–1194. [Google Scholar] [CrossRef] [Green Version]
- Schulz, K.F.; Altman, D.G.; Moher, D. CONSORT 2010 Statement: Updated Guidelines for Reporting Parallel Group Randomised Trials. BMJ 2010, 340, 698–702. [Google Scholar] [CrossRef]
- Prescott, M.J.; Lidster, K. Improving Quality of Science through Better Animal Welfare: The NC3Rs Strategy. Lab Anim. 2017, 46, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.L.; Trotman-Lucas, M.; Wong, R.; Allan, S.M. Improved Reperfusion Following Alternative Surgical Approach for Experimental Stroke in Mice. F1000Research 2020, 9, 188. [Google Scholar] [CrossRef]
- Macrae, I.M.; Allan, S.M. Stroke: The Past, Present and Future. Brain Neurosci. Adv. 2018, 2, 239821281881068. [Google Scholar] [CrossRef]
- Paul, S.; Candelario-Jalil, E. Emerging Neuroprotective Strategies for the Treatment of Ischemic Stroke: An Overview of Clinical and Preclinical Studies HHS Public Access. Exp. Neurol. 2021, 335, 113518. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Therapeutic Drug | Mechanism of Action | Status of Clinical Trial |
|---|---|---|---|
| VENUS (Phase III) | Nimodipine | Blocks voltage-gated channels (calcium) | Terminated: not effective |
| MAVARIC (Phase I) | Verapamil (with magnesium sulfate) | Blocks of voltage-gated channels (calcium), after reperfusion | Results not published yet |
| ESCAPE-NA1 (Phase III) | Nerinetide (Na-1) | Inhibits neuronal excitotoxicity | Approved by American FDA alone; but it does not display protection together with IVT |
| ESCAPE-NEXT (Phase III) | Nerinetide (Na-1) together with EVT therapy | Inhibits neuronal excitotoxicity | Ongoing (completes in August 2023) |
| URICO-ICTUS (Phase III) | Uric Acid (UA) | Antioxidant agent (reduces inflammation) | Small study sample, but a confirmatory trial is planned |
| BAST (Phase III) | 3-N-butylphtalide (NBP) | Free radical scavenger (reduces inflammation) | Ongoing (completed on 31 December 2022), good outcome so far |
| Evaluation of HUK in AIS (Phase IV) | Human Urinary Kallidinogenase (HUK) | Anti-inflammatory agents suppress TLR4/NF-kB pathway | Approved by China’s FDA, with post-approval surveillance ongoing |
| A study of allogeneic mesenchymal bone marrow cells in AIS (Phase I/II) | Allogeneic adult mesenchymal bone marrow cells | Cell-based therapy | Completed, presented beneficial behavioral outcomes for patients |
| J-REPAIR (Phase I/II) | JTR-161 (allogeneic stem cell product) | Cell-based therapy | Last updated on June 2022, results not published yet |
| Efficacy and safety of FTY720 for AIS (phase II) | Fingolimod (FTY720) | Immune modulator | Completed; reduced injury and improved clinical outcomes |
| Combination of the immune modulator Dimethyl Fumarate with alteplase in AIS (phase I/II) | Dimethyl Fumarate | Immune modulator | Ongoing (completes in December 2022) |
| Cellular Source | Culture Conditions | Advantages | Disadvantages | |
|---|---|---|---|---|
| Spheroids | Differentiated cells, multicellular mixture, primary cells | Cultures with or without ECM and growth factors, self-assemble on heterogenous sphere format |
|
|
| Organoids | Only stem cells: embryonic, adult, or pluripotent | Require ECM and growth factors, self-assemble with different cell lineages, resembling the structure and function of an organ |
|
|
| In vivo Stroke Model | Advantages | Disadvantages |
|---|---|---|
A. Intraluminal suture MCAO model |
|
|
B. Electrocoagulation of MCA model |
|
|
C. Embolic stroke model |
|
|
D. Endothelin-1 model |
|
|
E. Photothrombosis model |
|
|
 .
.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amado, B.; Melo, L.; Pinto, R.; Lobo, A.; Barros, P.; Gomes, J.R. Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models. Biomedicines 2022, 10, 2561. https://doi.org/10.3390/biomedicines10102561
Amado B, Melo L, Pinto R, Lobo A, Barros P, Gomes JR. Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models. Biomedicines. 2022; 10(10):2561. https://doi.org/10.3390/biomedicines10102561
Chicago/Turabian StyleAmado, Beatriz, Lúcia Melo, Raquel Pinto, Andrea Lobo, Pedro Barros, and João R. Gomes. 2022. "Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models" Biomedicines 10, no. 10: 2561. https://doi.org/10.3390/biomedicines10102561
APA StyleAmado, B., Melo, L., Pinto, R., Lobo, A., Barros, P., & Gomes, J. R. (2022). Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models. Biomedicines, 10(10), 2561. https://doi.org/10.3390/biomedicines10102561