
Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
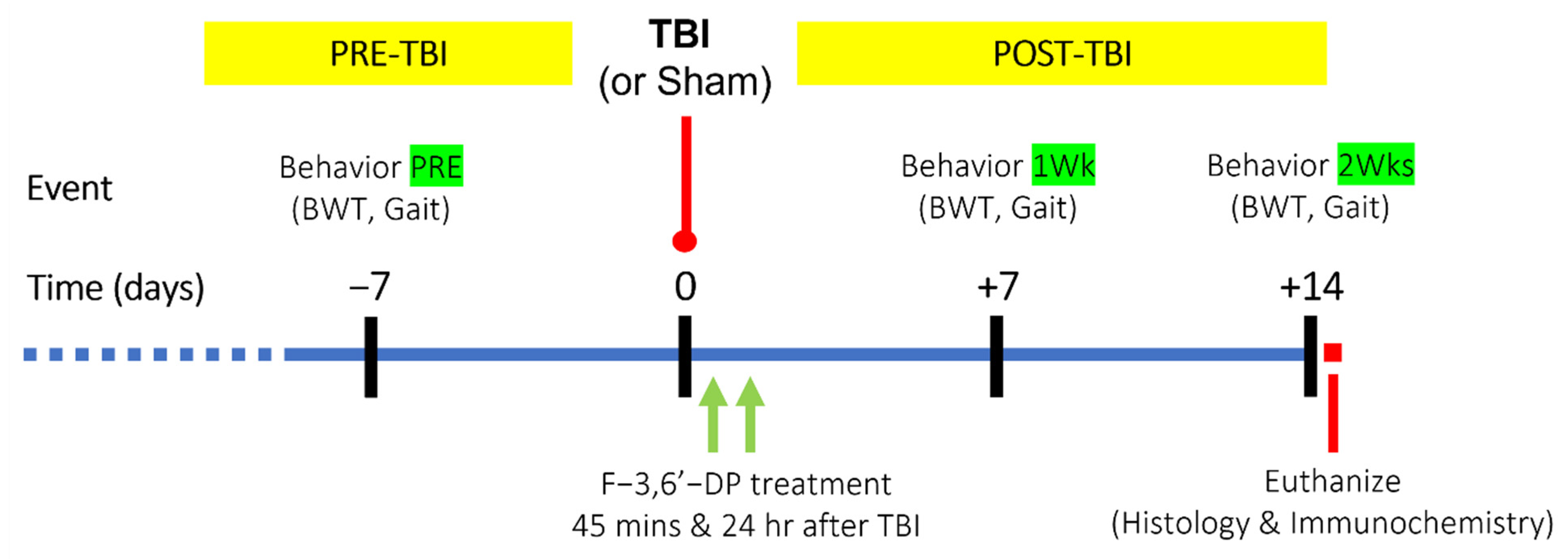
2.1. Animal Studies
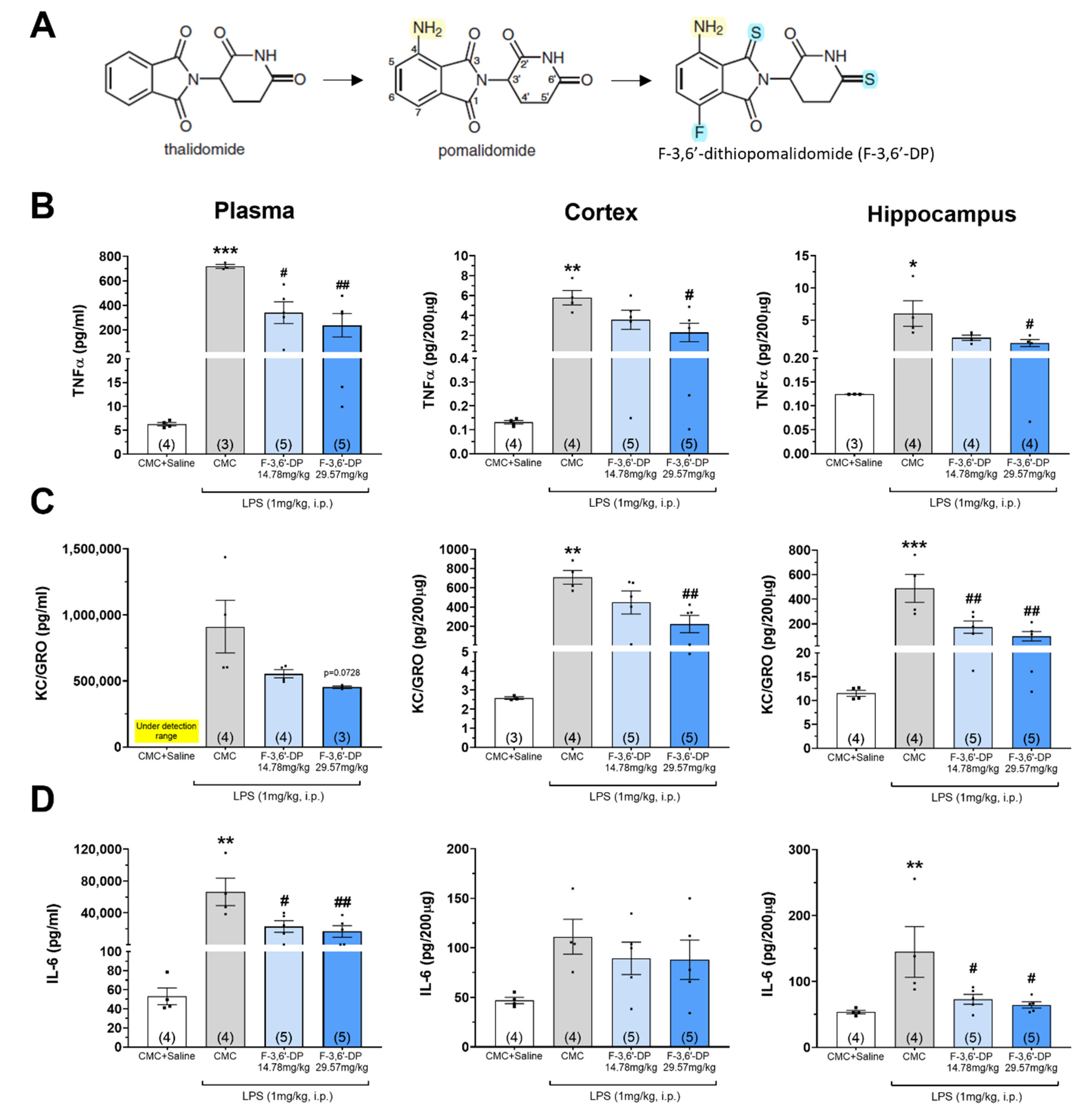
2.1.1. Synthesis and Analysis of F-3,6′-DP
Beam Walk Test
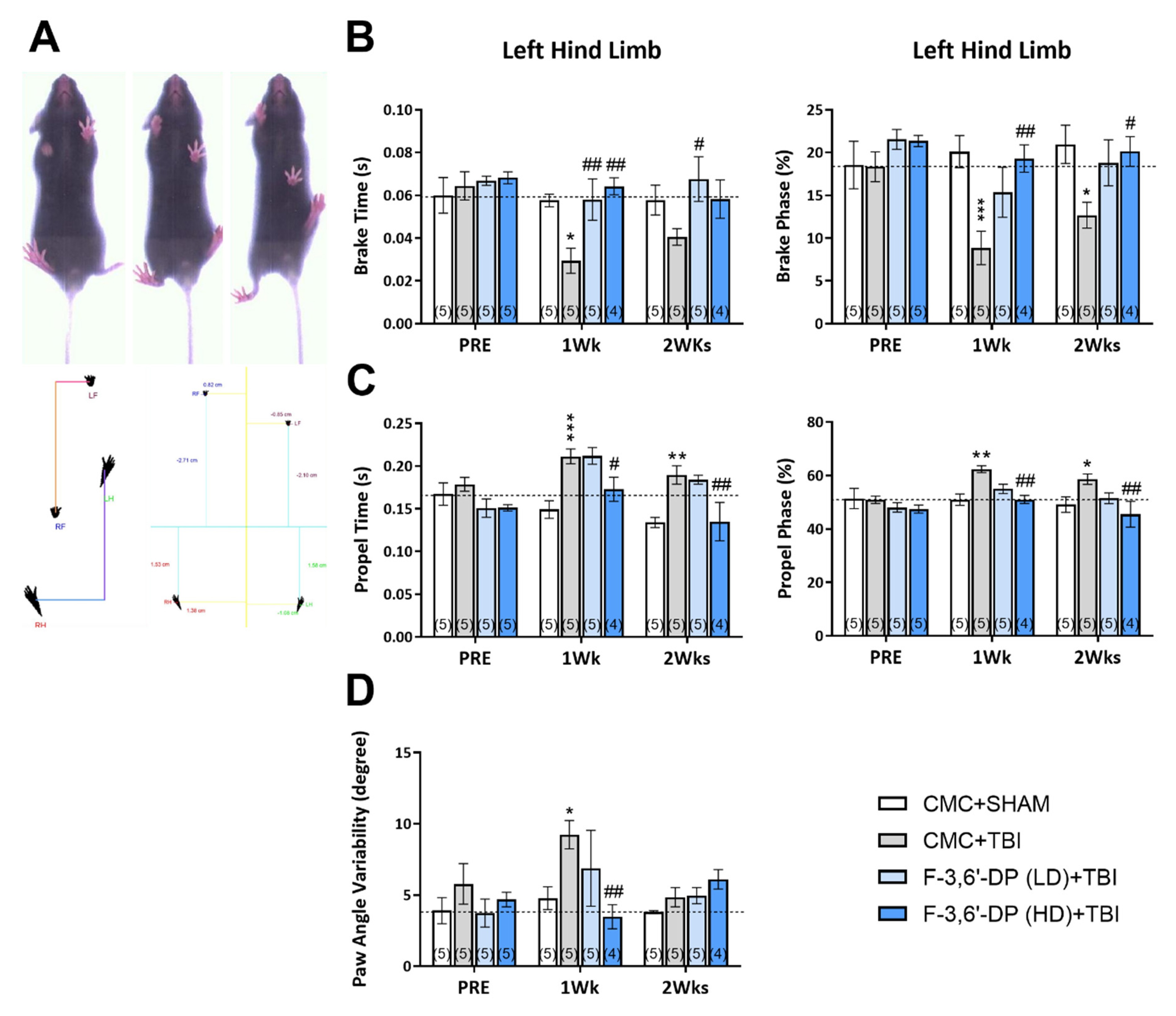
Gait Analysis
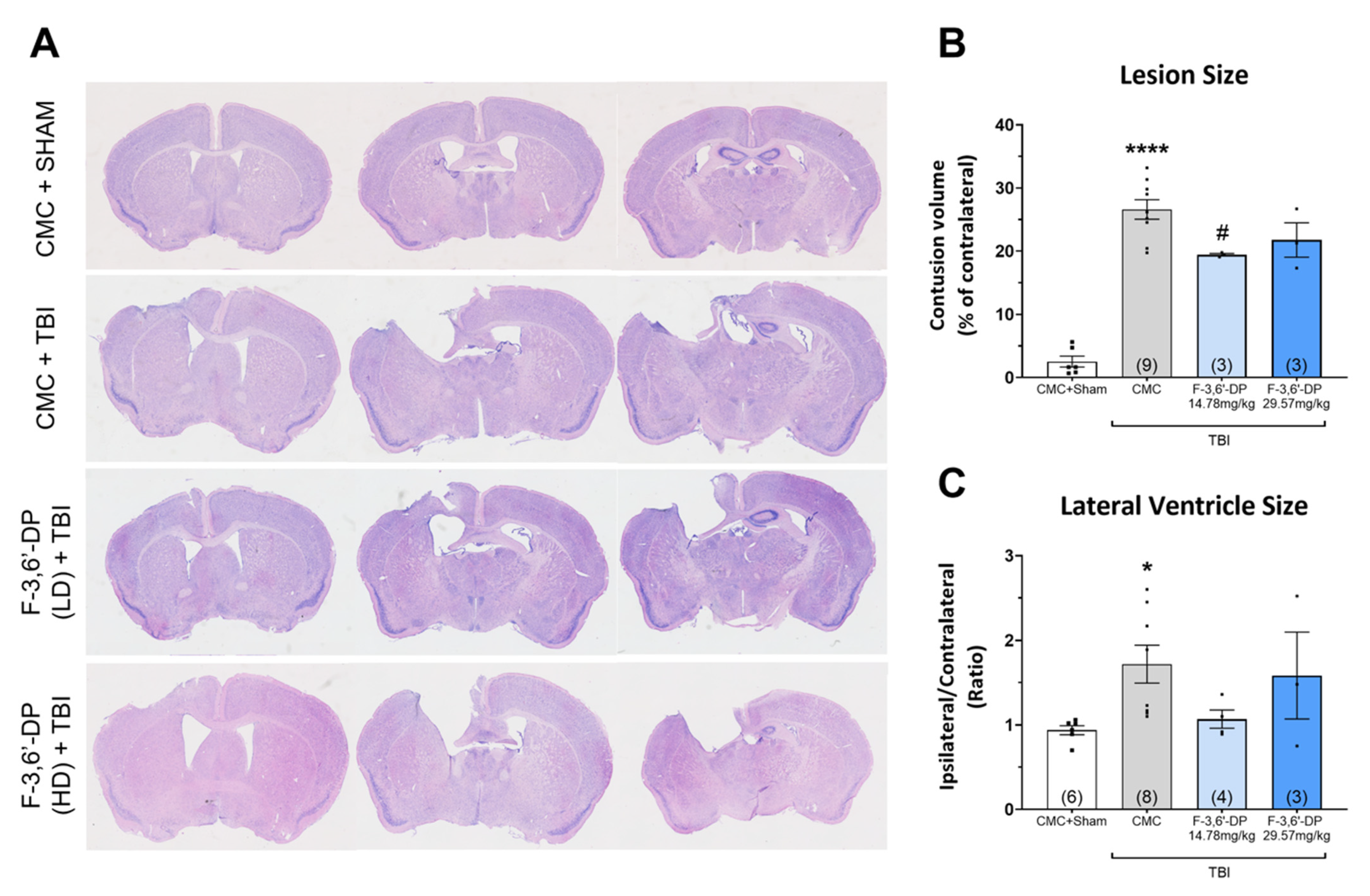
Histological Analysis
Immunofluorescence
2.1.2. Statistical Analysis
2.2. Cellular Studies
2.2.1. Cereblon Binding and Neo-Substrate Assays
2.2.2. Cellular Studies in RAW Cells
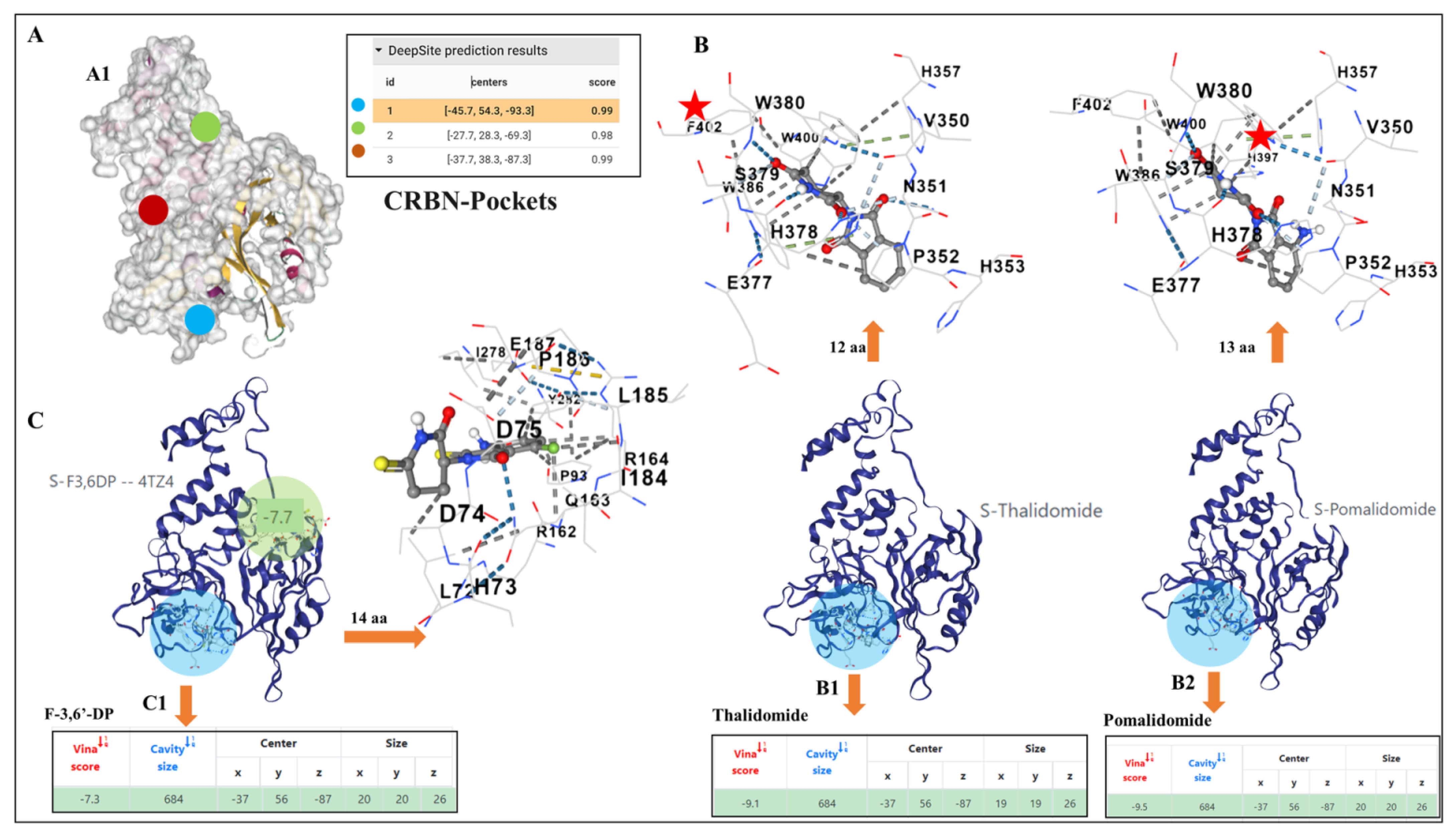
2.3. Docking Pockets and Predictions of F-3,6′-DP and Thalidomide Structural Analog Interactions with Cereblon
3. Results
3.1. Animal Studies
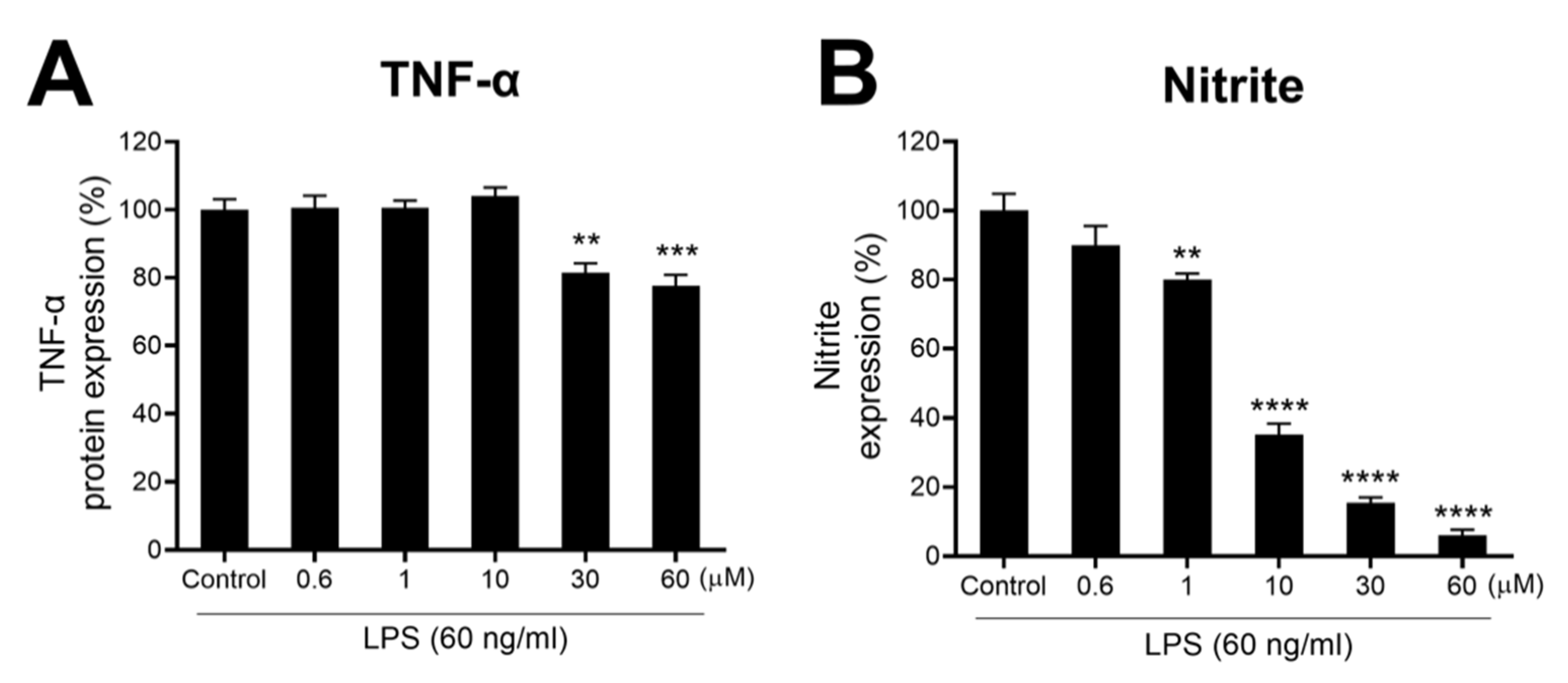
3.1.1. F-3,6′-DP Reduces the Expression of Pro-Inflammatory Cytokines, TNF-α, IL-6 and Chemokine KC/GRO (CXCL1) Induced by LPS in Rats
3.1.2. F-3,6′-DP Ameliorates Behavioral Deficits and Contusion Volume Produced by TBI
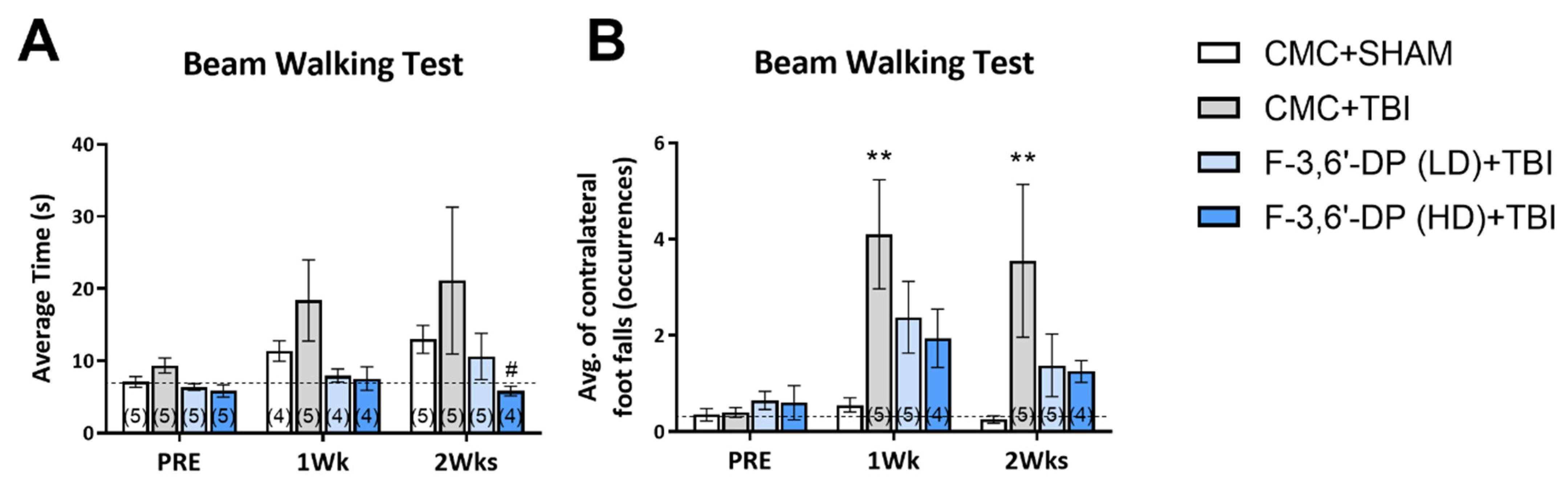
3.1.3. F-3,6′-DP Attenuates Gait Impairments Caused by TBI
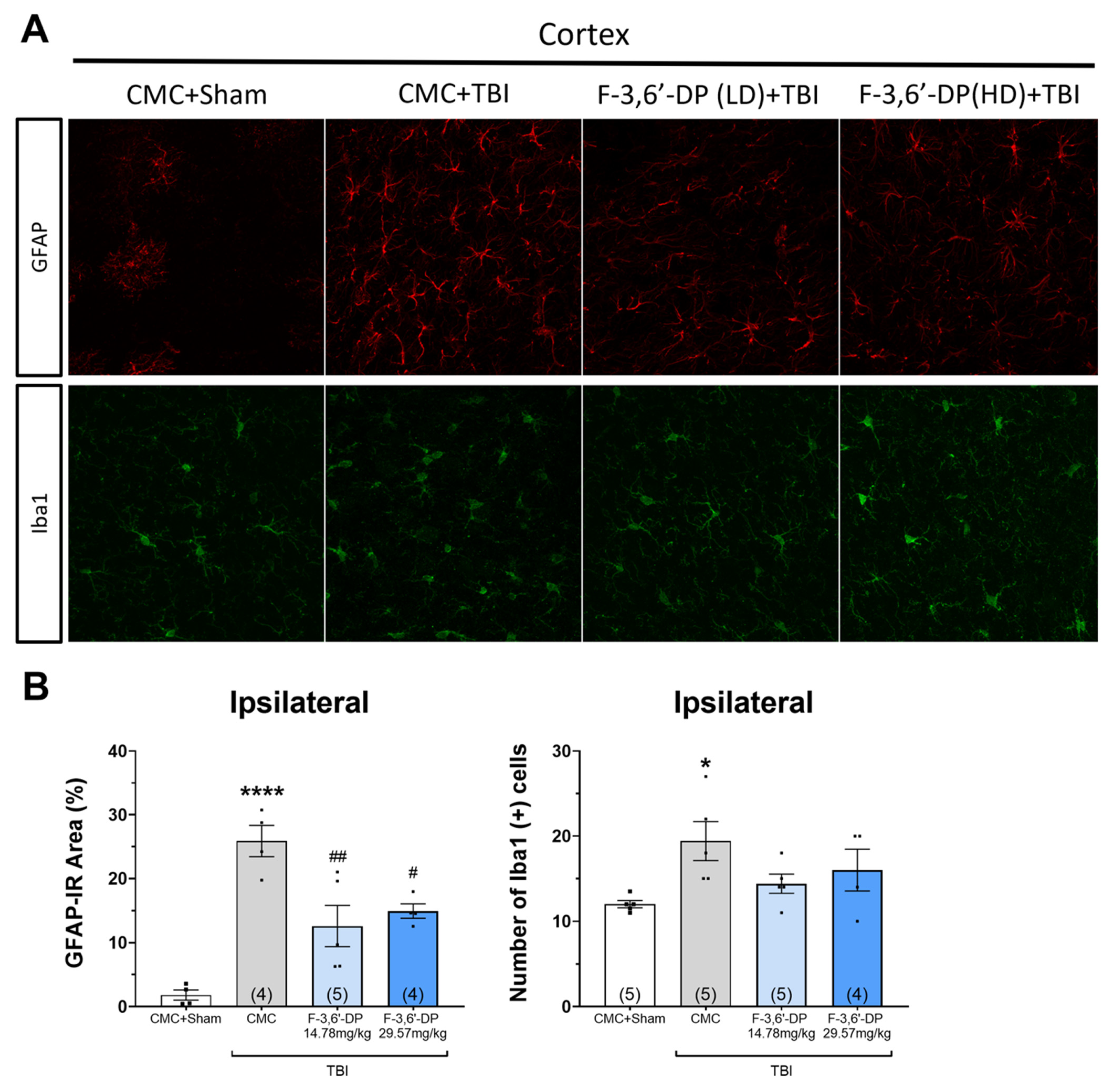
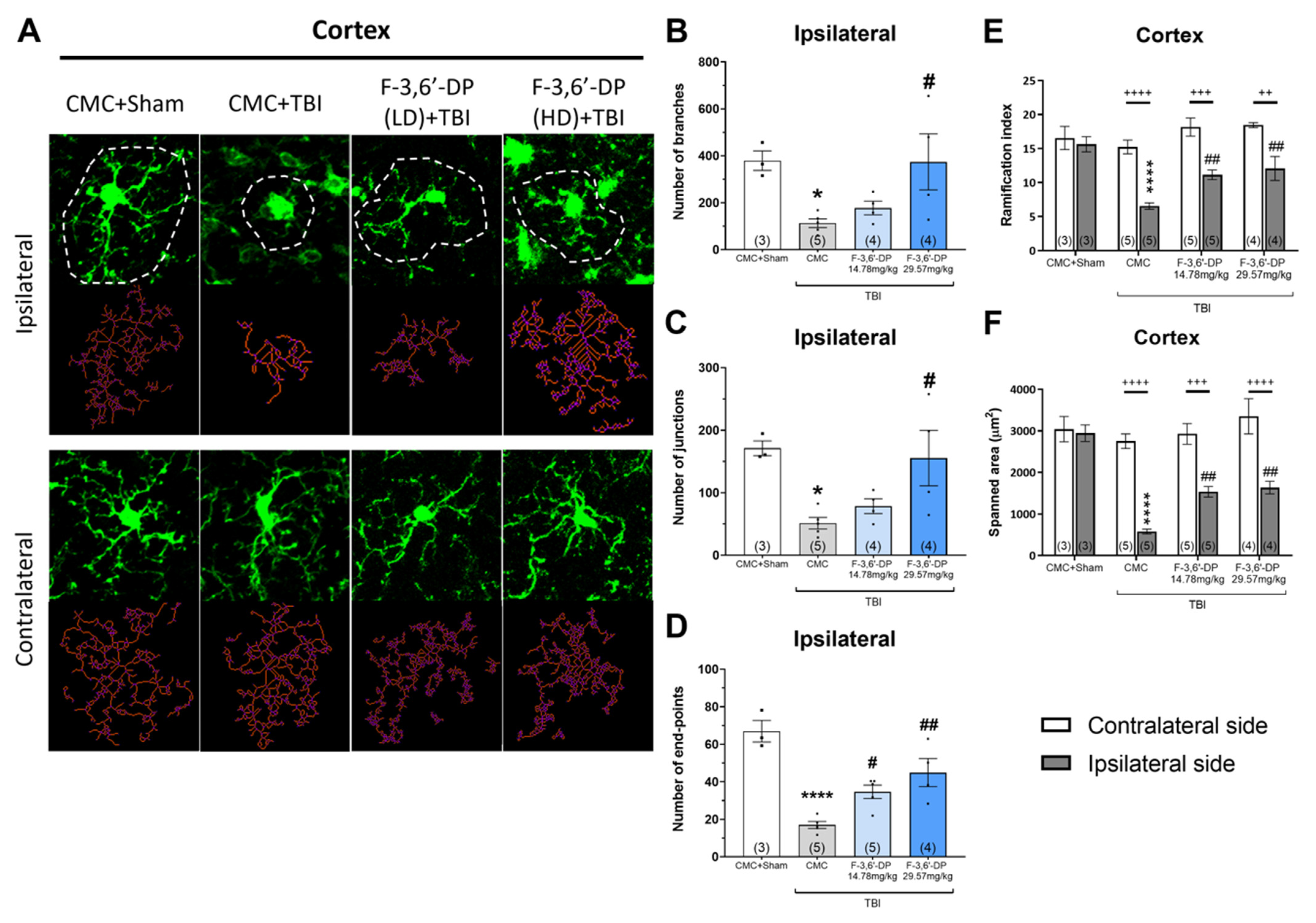
3.1.4. F-3,6′-DP Treatment Reduced the Activation of Microglia and Astrocytes Induced by TBI
3.2. Results: Cellular Studies
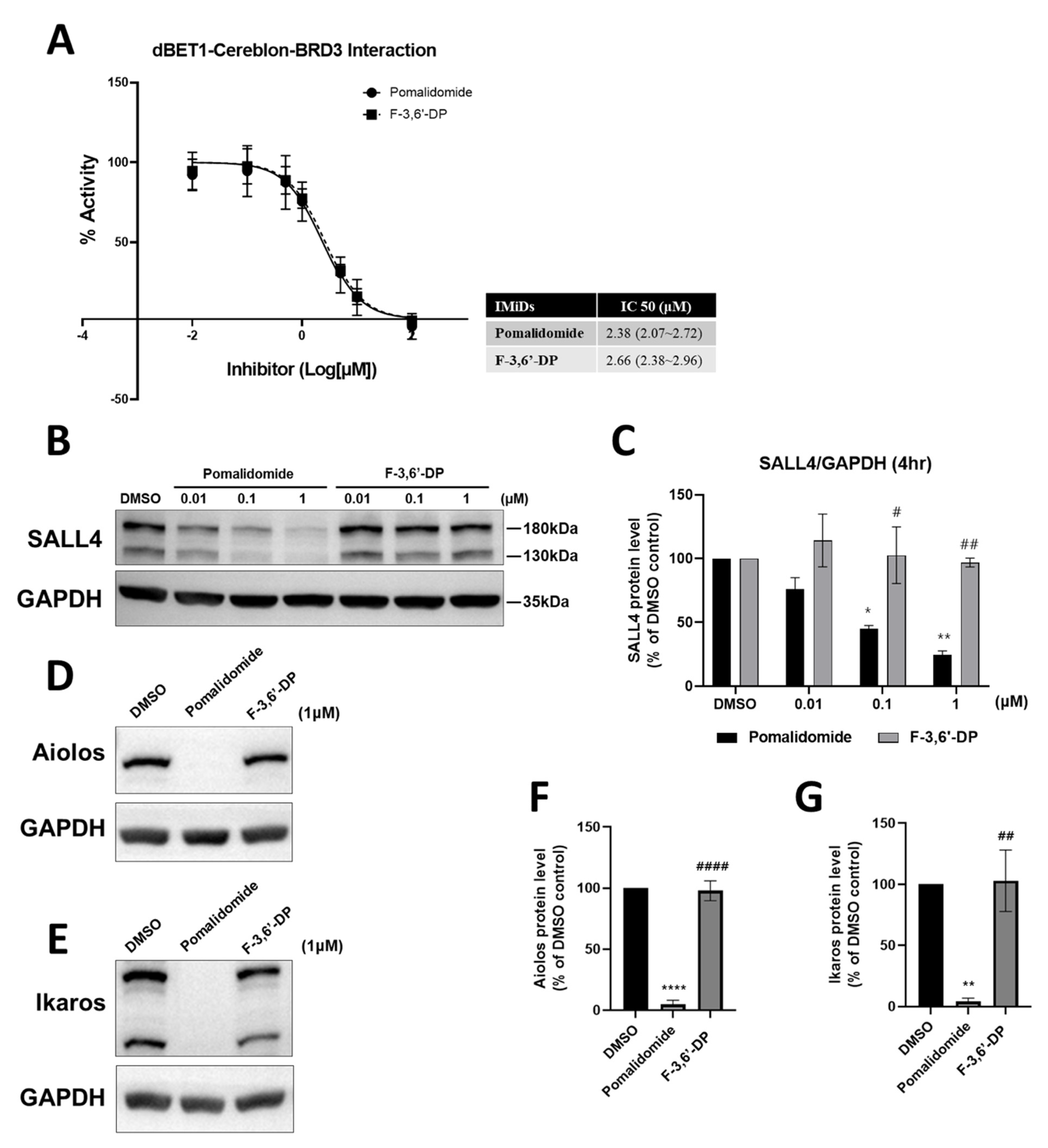
Cereblon Binding and Neo-Substrate Assays
3.3. Potential Differences and Similarities in the Docking Pocket for F-3,6′-DP, Thalidomide and Pomalidomide in Human Cereblon, as Evaluated by Molecular Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef]
- Archer, T. Influence of physical exercise on traumatic brain injury deficits: Scaffolding effect. Neurotox. Res. 2012, 21, 418–434. [Google Scholar] [CrossRef]
- Webb, N.E.; Little, B.; Loupee-Wilson, S.; Power, E.M. Traumatic brain injury and neuro-endocrine disruption: Medical and psychosocial rehabilitation. NeuroRehabilitation 2014, 34, 625–636. [Google Scholar] [CrossRef]
- Williams, G.; Morris, M.E.; Schache, A.; McCrory, P.R. Incidence of gait abnormalities after traumatic brain injury. Arch. Phys. Med. Rehabil. 2009, 90, 587–593. [Google Scholar] [CrossRef]
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association of Traumatic Brain Injury with Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol. 2016, 73, 1062–1069. [Google Scholar] [CrossRef]
- Shahaduzzaman, M.; Acosta, S.; Bickford, P.C.; Borlongan, C.V. alpha-Synuclein is a pathological link and therapeutic target for Parkinson’s disease and traumatic brain injury. Med. Hypotheses 2013, 81, 675–680. [Google Scholar] [CrossRef]
- Wong, J.C.; Hazrati, L.N. Parkinson’s disease, parkinsonism, and traumatic brain injury. Crit. Rev. Clin. Lab. Sci. 2013, 50, 103–106. [Google Scholar] [CrossRef]
- Fann, J.R.; Ribe, A.R.; Pedersen, H.S.; Fenger-Gron, M.; Christensen, J.; Benros, M.E.; Vestergaard, M. Long-term risk of dementia among people with traumatic brain injury in Denmark: A population-based observational cohort study. Lancet Psychiatr. 2018, 5, 424–431. [Google Scholar] [CrossRef]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, e1771–e1779. [Google Scholar] [CrossRef]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef]
- Cederberg, D.; Siesjo, P. What has inflammation to do with traumatic brain injury? Childs Nerv. Syst. 2010, 26, 221–226. [Google Scholar] [CrossRef]
- Utagawa, A.; Truettner, J.S.; Dietrich, W.D.; Bramlett, H.M. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp. Neurol. 2008, 211, 283–291. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Blennow, K.; Ikonomovic, M.D.; Gandy, S. Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2013, 9, 192–200. [Google Scholar] [CrossRef]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Webster, K.M.; Sun, M.; Crack, P.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflamm. 2017, 14, 10. [Google Scholar] [CrossRef]
- Chiu, C.C.; Liao, Y.E.; Yang, L.Y.; Wang, J.Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflamm. 2017, 2017, 8385961. [Google Scholar] [CrossRef]
- Nonaka, M.; Chen, X.H.; Pierce, J.E.; Leoni, M.J.; McIntosh, T.K.; Wolf, J.A.; Smith, D.H. Prolonged activation of NF-kappaB following traumatic brain injury in rats. J. Neurotrauma 1999, 16, 1023–1034. [Google Scholar] [CrossRef]
- Scherbel, U.; Raghupathi, R.; Nakamura, M.; Saatman, K.E.; Trojanowski, J.Q.; Neugebauer, E.; Marino, M.W.; McIntosh, T.K. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. USA 1999, 96, 8721–8726. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, E.R.; Prough, D.S. Interleukin-8, neuroinflammation, and secondary brain injury. Crit. Care Med. 2000, 28, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.; Morganti-Kossmann, M.C. The role of markers of inflammation in traumatic brain injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Dalgard, C.L.; Cole, J.T.; Kean, W.S.; Lucky, J.J.; Sukumar, G.; McMullen, D.C.; Pollard, H.B.; Watson, W.D. The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci. 2012, 5, 6. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Shohami, E.; Ginis, I.; Hallenbeck, J.M. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999, 10, 119–130. [Google Scholar] [CrossRef]
- Longhi, L.; Perego, C.; Ortolano, F.; Aresi, S.; Fumagalli, S.; Zanier, E.R.; Stocchetti, N.; De Simoni, M.G. Tumor necrosis factor in traumatic brain injury: Effects of genetic deletion of p55 or p75 receptor. J. Cereb. Blood Flow Metab. 2013, 33, 1182–1189. [Google Scholar] [CrossRef]
- Chang, R.; Yee, K.L.; Sumbria, R.K. Tumor necrosis factor alpha Inhibition for Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517709278. [Google Scholar] [CrossRef]
- Paouri, E.; Tzara, O.; Kartalou, G.I.; Zenelak, S.; Georgopoulos, S. Peripheral Tumor Necrosis Factor-Alpha (TNF-alpha) Modulates Amyloid Pathology by Regulating Blood-Derived Immune Cells and Glial Response in the Brain of AD/TNF Transgenic Mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Lindenau, J.D.; Altmann, V.; Schumacher-Schuh, A.F.; Rieder, C.R.; Hutz, M.H. Tumor necrosis factor alpha polymorphisms are associated with Parkinson’s disease age at onset. Neurosci. Lett. 2017, 658, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014, 20, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Scarlata, C.; Brambilla, L.; Rossi, D. Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells 2021, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Tortarolo, M.; Coco, D.L.; Veglianese, P.; Vallarola, A.; Giordana, M.T.; Marcon, G.; Beghi, E.; Poloni, M.; Strong, M.J.; Iyer, A.M.; et al. Amyotrophic Lateral Sclerosis, a Multisystem Pathology: Insights into the Role of TNFalpha. Mediat. Inflamm. 2017, 2017, 2985051. [Google Scholar] [CrossRef]
- Frugier, T.; Morganti-Kossmann, M.C.; O’Reilly, D.; McLean, C.A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 2010, 27, 497–507. [Google Scholar] [CrossRef]
- Csuka, E.; Morganti-Kossmann, M.C.; Lenzlinger, P.M.; Joller, H.; Trentz, O.; Kossmann, T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: Relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J. Neuroimmunol. 1999, 101, 211–221. [Google Scholar] [CrossRef]
- Riva-Depaty, I.; Fardeau, C.; Mariani, J.; Bouchaud, C.; Delhaye-Bouchaud, N. Contribution of peripheral macrophages and microglia to the cellular reaction after mechanical or neurotoxin-induced lesions of the rat brain. Exp. Neurol. 1994, 128, 77–87. [Google Scholar] [CrossRef]
- Shohami, E.; Gallily, R.; Mechoulam, R.; Bass, R.; Ben-Hur, T. Cytokine production in the brain following closed head injury: Dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J. Neuroimmunol. 1997, 72, 169–177. [Google Scholar] [CrossRef]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Longhi, L.; Ortolano, F.; Zanier, E.R.; Perego, C.; Stocchetti, N.; De Simoni, M.G. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir. Suppl. 2008, 102, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Berrrrpohl, D.; You, Z.; Lo, E.H.; Kim, H.H.; Whalen, M.J. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Somers, G.F. Pharmacological properties of thalidomide (alpha-phthalimido glutarimide), a new sedative hypnotic drug. Br. J. Pharmacol. Chemother. 1960, 15, 111–116. [Google Scholar] [CrossRef]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef]
- Rowland, T.L.; McHugh, S.M.; Deighton, J.; Ewan, P.W.; Dearman, R.J.; Kimber, I. Selective down-regulation of T cell- and non-T cell-derived tumour necrosis factor alpha by thalidomide: Comparisons with dexamethasone. Immunol. Lett. 1999, 68, 325–332. [Google Scholar] [CrossRef]
- Zhu, X.; Giordano, T.; Yu, Q.S.; Holloway, H.W.; Perry, T.A.; Lahiri, D.K.; Brossi, A.; Greig, N.H. Thiothalidomides: Novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J. Med. Chem. 2003, 46, 5222–5229. [Google Scholar] [CrossRef]
- McBride, W.G. Teratogenic action of thalidomide. Lancet 1978, 311, 1362. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef]
- Yang, T.J.; Yang, T.S.; Liang, H.M. Thalidomide and congenital abnormalities. Lancet 1963, 281, 552–553. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [PubMed]
- Zeldis, J.B.; Knight, R.; Hussein, M.; Chopra, R.; Muller, G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann. N. Y. Acad. Sci. 2011, 1222, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Guevara, P.; Tamariz, J.; Rembao, D.; Rivera, E.; Sotelo, J. Antiproliferative effect of thalidomide alone and combined with carmustine against C6 rat glioma. Int. J. Exp. Pathol. 2002, 83, 99–104. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Taylor, J.D.; Osiecki, L.L.; Kale-Pradhan, P.B. Novel therapies for Crohn’s disease: Focus on immunomodulators and antibiotics. Ann. Pharmacother. 2006, 40, 1804–1813. [Google Scholar] [CrossRef]
- Kurtin, S.E.; List, A.F. Durable long-term responses in patients with myelodysplastic syndromes treated with lenalidomide. Clin. Lymphoma Myeloma 2009, 9, E10–E13. [Google Scholar] [CrossRef]
- Shortt, J.; Hsu, A.K.; Johnstone, R.W. Thalidomide-analogue biology: Immunological, molecular and epigenetic targets in cancer therapy. Oncogene 2013, 32, 4191–4202. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Boichenko, I.; Bar, K.; Deiss, S.; Heim, C.; Albrecht, R.; Lupas, A.N.; Alvarez, B.H.; Hartmann, M.D. Chemical Ligand Space of Cereblon. ACS Omega 2018, 3, 11163–11171. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Cathers, B.E. Cereblon modulators: Low molecular weight inducers of protein degradation. Drug Discov. Today Technol. 2019, 31, 29–34. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Tweedie, D.; Beedie, S.L.; Vargesson, N.; Figg, W.D.; Greig, N.H.; Scerba, M.T. Design, synthesis and biological assessment of N-adamantyl, substituted adamantyl and noradamantyl phthalimidines for nitrite, TNF-alpha and angiogenesis inhibitory activities. Bioorg. Med. Chem. 2018, 26, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Kim, D.S.; Palmas, M.F.; Pisanu, A.; Carta, A.R.; Greig, N.H. Repurposing Immunomodulatory Imide Drugs (IMiDs) in Neuropsychiatric and Neurodegenerative Disorders. Front. Neurosci. 2021, 15, 656921. [Google Scholar] [CrossRef]
- Mahony, C.; Erskine, L.; Niven, J.; Greig, N.H.; Figg, W.D.; Vargesson, N. Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 12703–12708. [Google Scholar] [CrossRef]
- Hanaizi, Z.; Flores, B.; Hemmings, R.; Camarero, J.; Sancho-Lopez, A.; Salmonson, T.; Gisselbrecht, C.; Laane, E.; Pignatti, F. The European medicines agency review of pomalidomide in combination with low-dose dexamethasone for the treatment of adult patients with multiple myeloma: Summary of the scientific assessment of the committee for medicinal products for human use. Oncologist 2015, 20, 329–334. [Google Scholar] [CrossRef]
- Wang, J.Y.; Huang, Y.N.; Chiu, C.C.; Tweedie, D.; Luo, W.; Pick, C.G.; Chou, S.Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H.; et al. Pomalidomide mitigates neuronal loss, neuroinflammation, and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflamm. 2016, 13, 168. [Google Scholar] [CrossRef]
- Tsai, Y.R.; Tweedie, D.; Navas-Enamorado, I.; Scerba, M.T.; Chang, C.F.; Lai, J.H.; Wu, J.C.; Chen, Y.H.; Kang, S.J.; Hoffer, B.J.; et al. Pomalidomide Reduces Ischemic Brain Injury in Rodents. Cell Transpl. 2019, 28, 439–450. [Google Scholar] [CrossRef]
- Scerba, M.T.; Siegler, M.A.; Greig, N.H. Thionation of Aminophthalimide Hindered Carbonyl Groups and Application to the Synthesis of 3,6′-Dithionated Pomalidomides. Synlett 2021, 32, 917–922. [Google Scholar] [CrossRef]
- Kyzer, J.L.; Martens, M. Metabolism and Toxicity of Fluorine Compounds. Chem. Res. Toxicol. 2021, 34, 678–680. [Google Scholar] [CrossRef]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimers Dement. 2022; ahead of print. [Google Scholar] [CrossRef]
- Tsai, Y.R.; Kim, D.S.; Hsueh, S.C.; Chen, K.Y.; Wu, J.C.; Wang, J.Y.; Tsou, Y.S.; Hwang, I.; Kim, Y.K.; Gil, D.; et al. 3,6′-and 1,6′-Dithiopomalidomide Mitigate Ischemic Stroke in Rats and Blunt Inflammation. Pharmaceutics 2022, 14, 950. [Google Scholar] [CrossRef] [PubMed]
- Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular mechanisms of cereblon-based drugs. Pharmacol. Ther. 2019, 202, 132–139. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018, 14, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.T.; Lecca, D.; Yang, L.Y.; Luo, W.; Scerba, M.T.; Tweedie, D.; Huang, P.S.; Jung, Y.J.; Kim, D.S.; Yang, C.H.; et al. 3,6’-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. eLife 2020, 9, e54726. [Google Scholar] [CrossRef]
- Martinez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- Jimenez, J.; Doerr, S.; Martinez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.T.; Hou, M.C.; Xiao, Z.X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Griffin, W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J. Neuroinflamm. 2016, 13, 236. [Google Scholar] [CrossRef] [PubMed]
- Degan, D.; Ornello, R.; Tiseo, C.; Carolei, A.; Sacco, S.; Pistoia, F. The Role of Inflammation in Neurological Disorders. Curr. Pharm. Des. 2018, 24, 1485–1501. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. Non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory agents in the treatment of neurodegenerative disease. Curr. Alzheimer Res. 2005, 2, 355–365. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis from Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef]
- Gyengesi, E.; Munch, G. In search of an anti-inflammatory drug for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 131–132. [Google Scholar] [CrossRef]
- Stein, D.G.; Sayeed, I. Repurposing and repositioning neurosteroids in the treatment of traumatic brain injury: A report from the trenches. Neuropharmacology 2019, 147, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Pattinson, C.L.; Guedes, V.A.; Peyer, J.; Moore, C.; Davis, T.; Devoto, C.; Turtzo, L.C.; Latour, L.; Gill, J.M. Inflammatory Cytokines Associate with Neuroimaging After Acute Mild Traumatic Brain Injury. Front. Neurol. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bai, L.; Niu, X.; Wang, Z.; Yin, B.; Bai, G.; Zhang, D.; Gan, S.; Sun, C.; Wang, S.; et al. Elevated Serum Levels of Inflammation-Related Cytokines in Mild Traumatic Brain Injury Are Associated with Cognitive Performance. Front. Neurol. 2019, 10, 1120. [Google Scholar] [CrossRef] [PubMed]
- Spittau, B. Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. The meteorology of cytokine storms, and the clinical usefulness of this knowledge. Semin. Immunopathol. 2017, 39, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, S.M.; Fan, L.; Faden, A.I. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J. Neuroimmunol. 1999, 95, 115–125. [Google Scholar] [CrossRef]
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 2018, 141, 459–471. [Google Scholar] [CrossRef]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Hill, D.; Guo, L.; Nicholas, R.; Papadopoulos, D.; Cordeiro, M.F. Automated characterisation of microglia in ageing mice using image processing and supervised machine learning algorithms. Sci. Rep. 2022, 12, 1806. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Jiang, T.; Telu, S.; Zoghbi, S.S.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Guo, Q.; Pike, V.W.; Innis, R.B.; et al. (11)C-DPA-713 has much greater specific binding to translocator protein 18 kDa (TSPO) in human brain than (11)C-(R)-PK11195. J. Cereb. Blood Flow Metab. 2018, 38, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Kumar, A.; Muzik, O.; Shandal, V.; Chugani, D.; Chakraborty, P.; Chugani, H.T. Evaluation of age-related changes in translocator protein (TSPO) in human brain using (11)C-[R]-PK11195 PET. J. Neuroinflamm. 2012, 9, 232. [Google Scholar] [CrossRef]
- George, K.; Das, J.M. Neuroanatomy, Thalamocortical Radiations; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Dever, A.; Powell, D.; Graham, L.; Mason, R.; Das, J.; Marshall, S.J.; Vitorio, R.; Godfrey, A.; Stuart, S. Gait Impairment in Traumatic Brain Injury: A Systematic Review. Sensors 2022, 22, 1480. [Google Scholar] [CrossRef]
- Williams, G.; Lai, D.; Schache, A.; Morris, M.E. Classification of gait disorders following traumatic brain injury. J. Head Trauma Rehabil. 2015, 30, E13–E23. [Google Scholar] [CrossRef]
- Osier, N.D.; Korpon, J.R.; Dixon, C.E. Controlled Cortical Impact Model. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2015. [Google Scholar]
- Yu, S.; Kaneko, Y.; Bae, E.; Stahl, C.E.; Wang, Y.; van Loveren, H.; Sanberg, P.R.; Borlongan, C.V. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009, 1287, 157–163. [Google Scholar] [CrossRef]
- Reed, J.; Grillakis, A.; Kline, A.; Ahmed, A.E.; Byrnes, K.R. Gait analysis in a rat model of traumatic brain injury. Behav. Brain Res. 2021, 405, 113210. [Google Scholar] [CrossRef]
- Sashindranath, M.; Daglas, M.; Medcalf, R.L. Evaluation of gait impairment in mice subjected to craniotomy and traumatic brain injury. Behav. Brain Res. 2015, 286, 33–38. [Google Scholar] [CrossRef]
- Cole, J.T.; Yarnell, A.; Kean, W.S.; Gold, E.; Lewis, B.; Ren, M.; McMullen, D.C.; Jacobowitz, D.M.; Pollard, H.B.; O’Neill, J.T.; et al. Craniotomy: True sham for traumatic brain injury, or a sham of a sham? J. Neurotrauma 2011, 28, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Lagraoui, M.; Latoche, J.R.; Cartwright, N.G.; Sukumar, G.; Dalgard, C.L.; Schaefer, B.C. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Front. Neurol. 2012, 3, 155. [Google Scholar] [CrossRef] [PubMed]
- Frigon, A. The neural control of interlimb coordination during mammalian locomotion. J. Neurophysiol. 2017, 117, 2224–2241. [Google Scholar] [CrossRef]
- Jacobowitz, D.M.; Cole, J.T.; McDaniel, D.P.; Pollard, H.B.; Watson, W.D. Microglia activation along the corticospinal tract following traumatic brain injury in the rat: A neuroanatomical study. Brain Res. 2012, 1465, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Batsaikhan, B.; Wang, J.Y.; Scerba, M.T.; Tweedie, D.; Greig, N.H.; Miller, J.P.; Hoffer, B.J.; Lin, C.T.; Wang, J.Y. Post-Injury Neuroprotective Effects of the Thalidomide Analog 3,6’-Dithiothalidomide on Traumatic Brain Injury. Int. J. Mol. Sci. 2019, 20, 502. [Google Scholar] [CrossRef] [PubMed]
- Baratz, R.; Tweedie, D.; Wang, J.Y.; Rubovitch, V.; Luo, W.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Transiently lowering tumor necrosis factor-alpha synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflamm. 2015, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ito, T.; Handa, H. Cereblon-based small-molecule compounds to control neural stem cell proliferation in regenerative medicine. Front. Cell Dev. Biol. 2021, 9, 629326. [Google Scholar] [CrossRef]
- Xu, G.; Jiang, X.; Jaffrey, S.R. A mental retardation-linked nonsense mutation in cereblon is rescued by proteasome inhibition. J. Biol. Chem. 2013, 288, 29573–29585. [Google Scholar] [CrossRef]
- Ito, T.; Handa, H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int. J. Hematol. 2016, 104, 293–299. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, L. Cereblon: A Protein Crucial to the Multiple Functions of Immunomodulatory Drugs as well as Cell Metabolism and Disease Generation. J. Immunol. Res. 2017, 2017, 9130608. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Matyskiela, M.E.; Clayton, T.; Zheng, X.; Mayne, C.; Tran, E.; Carpenter, A.; Pagarigan, B.; McDonald, J.; Rolfe, M.; Hamann, L.G.; et al. Crystal structure of the SALL4-pomalidomide-cereblon-DDB1 complex. Nat. Struct. Mol. Biol. 2020, 27, 319–322. [Google Scholar] [CrossRef]
- Gemechu, Y.; Millrine, D.; Hashimoto, S.; Prakash, J.; Sanchenkova, K.; Metwally, H.; Gyanu, P.; Kang, S.; Kishimoto, T. Humanized cereblon mice revealed two distinct therapeutic pathways of immunomodulatory drugs. Proc. Natl. Acad. Sci. USA 2018, 115, 11802–11807. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, M.; Zhou, L.; He, X.; Jiang, X.; Zhang, Y.; Xu, G. Cereblon suppresses the lipopolysaccharide-induced inflammatory response by promoting the ubiquitination and degradation of c-Jun. J. Biol. Chem. 2018, 293, 10141–10157. [Google Scholar] [CrossRef]
- Fink, E.C.; McConkey, M.; Adams, D.N.; Haldar, S.D.; Kennedy, J.A.; Guirguis, A.A.; Udeshi, N.D.; Mani, D.R.; Chen, M.; Liddicoat, B.; et al. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 2018, 132, 1535–1544. [Google Scholar] [CrossRef]
- Min, Y.; Wi, S.M.; Kang, J.A.; Yang, T.; Park, C.S.; Park, S.G.; Chung, S.; Shim, J.H.; Chun, E.; Lee, K.Y. Cereblon negatively regulates TLR4 signaling through the attenuation of ubiquitination of TRAF6. Cell Death Dis. 2016, 7, e2313. [Google Scholar] [CrossRef]
- Hsueh, S.C.; Luo, W.; Tweedie, D.; Kim, D.S.; Kim, Y.K.; Hwang, I.; Gil, J.E.; Han, B.S.; Chiang, Y.H.; Selman, W.; et al. N-Adamantyl phthalimidine: A new thalidomide-like drug that lacks cereblon binding and mitigates neuronal and synaptic loss, neuroinflammation, and behavioral deficits in traumatic brain injury and LPS challenge. ACS Pharmacol. Transl. Sci. 2021, 4, 980–1000. [Google Scholar] [CrossRef]
- Vargesson, N. The teratogenic effects of thalidomide on limbs. J. Hand Surg. 2019, 44, 88–95. [Google Scholar] [CrossRef]
- Gupte, R.; Brooks, W.; Vukas, R.; Pierce, J.; Harris, J. Sex differences in traumatic brain injury: What we know and what we should know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of estrogen and other sex hormones in brain aging. neuroprotection and DNA repair. Front. Aging Neurosci. 2017, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Soldin, O.P.; Mattison, D.R. Sex differences in pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex Differ. 2020, 11, 32. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsueh, S.C.; Scerba, M.T.; Tweedie, D.; Lecca, D.; Kim, D.S.; Baig, A.M.; Kim, Y.K.; Hwang, I.; Kim, S.; Selman, W.R.; et al. Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury. Biomedicines 2022, 10, 2449. https://doi.org/10.3390/biomedicines10102449
Hsueh SC, Scerba MT, Tweedie D, Lecca D, Kim DS, Baig AM, Kim YK, Hwang I, Kim S, Selman WR, et al. Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury. Biomedicines. 2022; 10(10):2449. https://doi.org/10.3390/biomedicines10102449
Chicago/Turabian StyleHsueh, Shih Chang, Michael T. Scerba, David Tweedie, Daniela Lecca, Dong Seok Kim, Abdul Mannan Baig, Yu Kyung Kim, Inho Hwang, Sun Kim, Warren R. Selman, and et al. 2022. "Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury" Biomedicines 10, no. 10: 2449. https://doi.org/10.3390/biomedicines10102449
APA StyleHsueh, S. C., Scerba, M. T., Tweedie, D., Lecca, D., Kim, D. S., Baig, A. M., Kim, Y. K., Hwang, I., Kim, S., Selman, W. R., Hoffer, B. J., & Greig, N. H. (2022). Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury. Biomedicines, 10(10), 2449. https://doi.org/10.3390/biomedicines10102449