

NF-κB Mutations in Germinal Center B-Cell Lymphomas: Relation to NF-κB Function in Normal B Cells
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
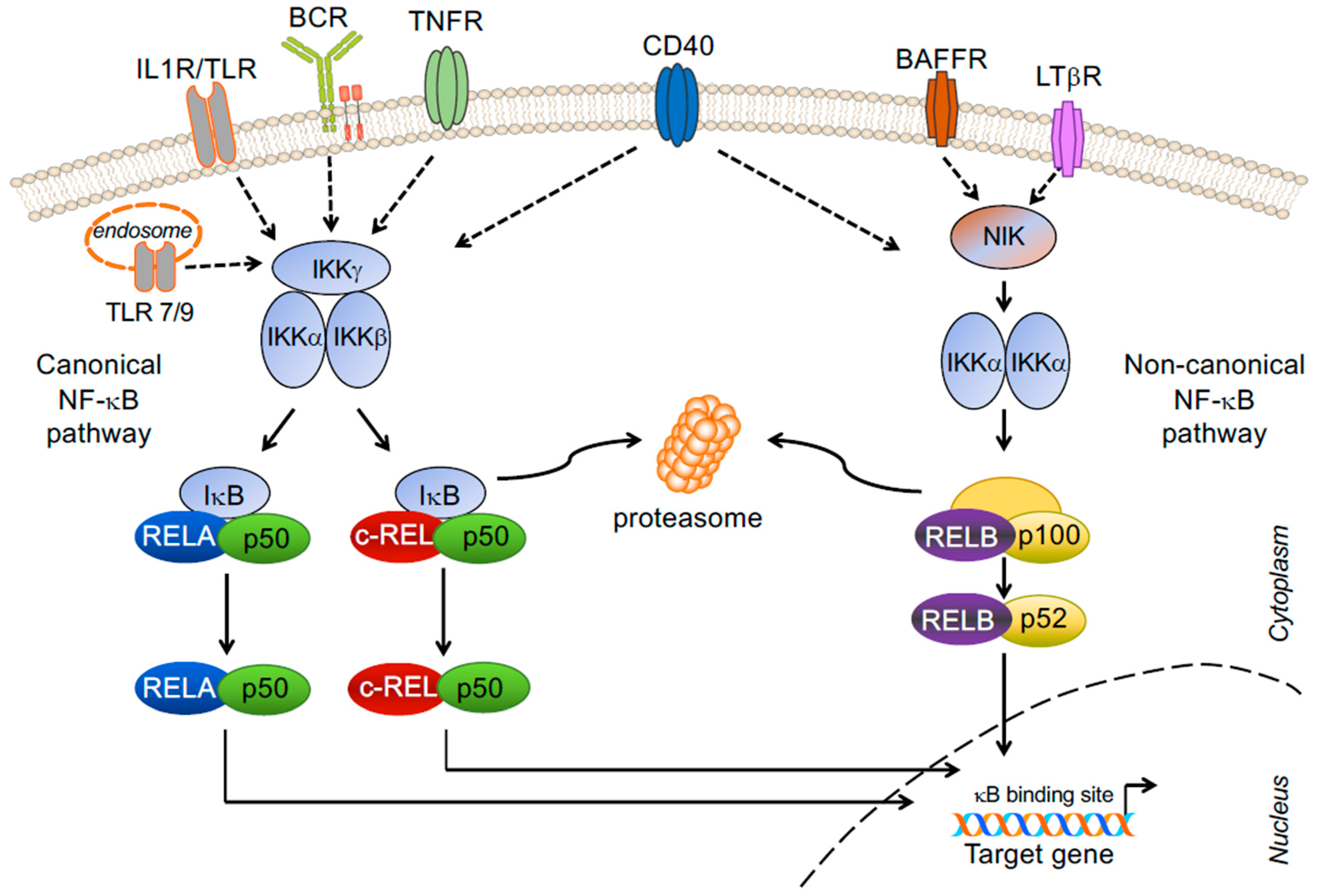
2. NF-κB Signaling Pathway and Transcription Factor Subunits
3. Role of NF-κB Signaling Pathways and Subunits in GC B-Cell Development
3.1. The Germinal Center B-Cell Reaction
3.2. NF-κB Signaling in the GC Reaction
3.3. Canonical Subunit c-REL
3.4. Canonical Subunit NF-ĸB1
3.5. Canonical Subunit RELA
3.6. Non-Canonical Subunits RELB and NF-ĸB2
4. Dysregulation of the NF-κB Signaling Pathway in DLBCL
4.1. Cell of Origin and Classifications
4.2. Mutations Leading to the Activation of the Canonical NF-κB Pathway
4.3. Genetic Lesions Leading to Activation of the Non-Canonical NF-κB Pathway
4.4. Potential Role of Downstream NF-κB Transcription Factors in DLBCL Lymphomagenesis
5. Hodgkin Lymphoma
6. Primary Mediastinal Large B-Cell Lymphoma
7. Therapeutic Implications
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Weniger, M.A.; Küppers, R. NF-kappaB deregulation in Hodgkin lymphoma. Semin. Cancer Biol. 2016, 39, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Chang, C.C.; Lombardi, L.; Salina, M.; Corradini, P.; Maiolo, A.T.; Chaganti, R.S.; Dalla-Favera, R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kappa B p50. Cell 1991, 67, 1075–1087. [Google Scholar] [CrossRef]
- Lu, D.; Thompson, J.D.; Gorski, G.K.; Rice, N.R.; Mayer, M.G.; Yunis, J.J. Alterations at the rel locus in human lymphoma. Oncogene 1991, 6, 1235–1241. [Google Scholar] [PubMed]
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene 1999, 18, 3063–3070. [Google Scholar] [CrossRef]
- Emmerich, F.; Meiser, M.; Hummel, M.; Demel, G.; Foss, H.D.; Jundt, F.; Mathas, S.; Krappmann, D.; Scheidereit, C.; Stein, H.; et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood 1999, 94, 3129–3134. [Google Scholar] [CrossRef]
- Jungnickel, B.; Staratschek-Jox, A.; Brauninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.L.; Rajewsky, K.; Küppers, R. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin’s lymphoma. J. Exp. Med. 2000, 191, 395–402. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef]
- Allen, C.D.; Okada, T.; Cyster, J.G. Germinal-center organization and cellular dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef]
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol 2017, 45, 21–30. [Google Scholar] [CrossRef]
- Küppers, R.; Klein, U.; Hansmann, M.L.; Rajewsky, K. Cellular origin of human B-cell lymphomas. N. Engl. J. Med. 1999, 341, 1520–1529. [Google Scholar] [CrossRef]
- Hauser, A.E.; Shlomchik, M.J.; Haberman, A.M. In vivo imaging studies shed light on germinal-centre development. Nat. Rev. Immunol. 2007, 7, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Dominguez-Sola, D.; Holmes, A.B.; Deroubaix, S.; Dalla-Favera, R.; Nussenzweig, M.C. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 2012, 120, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2022, 40, 413–442. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Mlynarczyk, C.; Fontan, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef]
- Pasqualucci, L. Molecular pathogenesis of germinal center-derived B cell lymphomas. Immunol. Rev. 2019, 288, 240–261. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L., 3rd; Young, R.M.; Staudt, L.M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef]
- Kaileh, M.; Sen, R. NF-kappaB function in B lymphocytes. Immunol. Rev. 2012, 246, 254–271. [Google Scholar] [CrossRef]
- Malinin, N.L.; Boldin, M.P.; Kovalenko, A.V.; Wallach, D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature 1997, 385, 540–544. [Google Scholar] [CrossRef]
- Woronicz, J.D.; Gao, X.; Cao, Z.; Rothe, M.; Goeddel, D.V. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science 1997, 278, 866–869. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Durandy, A.; Kracker, S.; Fischer, A. Primary antibody deficiencies. Nat. Rev. Immunol. 2013, 13, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M.J.; Luo, W.; Weisel, F. Linking signaling and selection in the germinal center. Immunol. Rev. 2019, 288, 49–63. [Google Scholar] [CrossRef]
- Basso, K.; Klein, U.; Niu, H.; Stolovitzky, G.A.; Tu, Y.; Califano, A.; Cattoretti, G.; Dalla-Favera, R. Tracking CD40 signaling during germinal center development. Blood 2004, 104, 4088–4096. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Rosenwald, A.; Hurt, E.M.; Giltnane, J.M.; Lam, L.T.; Pickeral, O.K.; Staudt, L.M. Signatures of the immune response. Immunity 2001, 15, 375–385. [Google Scholar] [CrossRef]
- De Silva, N.S.; Anderson, M.M.; Carette, A.; Silva, K.; Heise, N.; Bhagat, G.; Klein, U. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 9063–9068. [Google Scholar] [CrossRef]
- Kontgen, F.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Li, R.; Tarlinton, D.; Gerondakis, S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995, 9, 1965–1977. [Google Scholar] [CrossRef]
- Pohl, T.; Gugasyan, R.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Tarlinton, D.; Sha, W.; Baltimore, D.; Gerondakis, S. The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc. Natl. Acad. Sci. USA 2002, 99, 4514–4519. [Google Scholar] [CrossRef]
- Tumang, J.R.; Owyang, A.; Andjelic, S.; Jin, Z.; Hardy, R.R.; Liou, M.L.; Liou, H.C. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur. J. Immunol. 1998, 28, 4299–4312. [Google Scholar] [CrossRef]
- Milanovic, M.; Heise, N.; De Silva, N.S.; Anderson, M.M.; Silva, K.; Carette, A.; Orelli, F.; Bhagat, G.; Klein, U. Differential requirements for the canonical NF-kappaB transcription factors c-REL and RELA during the generation and activation of mature B cells. Immunol. Cell Biol. 2017, 95, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; De Silva, N.S.; Silva, K.; Carette, A.; Simonetti, G.; Pasparakis, M.; Klein, U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J. Exp. Med. 2014, 211, 2103–2118. [Google Scholar] [CrossRef] [PubMed]
- Calado, D.P.; Sasaki, Y.; Godinho, S.A.; Pellerin, A.; Kochert, K.; Sleckman, B.P.; de Alboran, I.M.; Janz, M.; Rodig, S.; Rajewsky, K. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 2012, 13, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef]
- Luo, W.; Weisel, F.; Shlomchik, M.J. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 2018, 48, 313–326.e5. [Google Scholar] [CrossRef]
- Finkin, S.; Hartweger, H.; Oliveira, T.Y.; Kara, E.E.; Nussenzweig, M.C. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity 2019, 51, 324–336.e5. [Google Scholar] [CrossRef]
- Gitlin, A.D.; Shulman, Z.; Nussenzweig, M.C. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 2014, 509, 637–640. [Google Scholar] [CrossRef]
- Kennedy, D.E.; Okoreeh, M.K.; Maienschein-Cline, M.; Ai, J.; Veselits, M.; McLean, K.C.; Dhungana, Y.; Wang, H.; Peng, J.; Chi, H.; et al. Novel specialized cell state and spatial compartments within the germinal center. Nat. Immunol. 2020, 21, 660–670. [Google Scholar] [CrossRef]
- Kober-Hasslacher, M.; Oh-Strauss, H.; Kumar, D.; Soberon, V.; Diehl, C.; Lech, M.; Engleitner, T.; Katab, E.; Fernandez-Saiz, V.; Piontek, G.; et al. c-Rel gain in B cells drives germinal center reactions and autoantibody production. J. Clin. Investig. 2020, 130, 3270–3286. [Google Scholar] [CrossRef]
- Beaussant-Cohen, S.; Jaber, F.; Massaad, M.J.; Weeks, S.; Jones, J.; Alosaimi, M.F.; Wallace, J.; Al-Herz, W.; Geha, R.S.; Chou, J. Combined immunodeficiency in a patient with c-Rel deficiency. J. Allergy Clin. Immunol. 2019, 144, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Langlais, D.; Beziat, V.; Rapaport, F.; Rao, G.; Lazarov, T.; Bourgey, M.; Zhou, Y.J.; Briand, C.; Moriya, K.; et al. Inherited human c-Rel deficiency disrupts myeloid and lymphoid immunity to multiple infectious agents. J. Clin. Investig. 2021, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Schweighoffer, E.; Visekruna, A.; Papoutsopoulou, S.; Janzen, J.; Zillwood, R.; Tarlinton, D.M.; Tybulewicz, V.L.; Ley, S.C. IKK-induced NF-kappaB1 p105 proteolysis is critical for B cell antibody responses to T cell-dependent antigen. J. Exp. Med. 2014, 211, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- de Valle, E.; Grigoriadis, G.; O’Reilly, L.A.; Willis, S.N.; Maxwell, M.J.; Corcoran, L.M.; Tsantikos, E.; Cornish, J.K.; Fairfax, K.A.; Vasanthakumar, A.; et al. NFkappaB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J. Exp. Med. 2016, 213, 621–641. [Google Scholar] [CrossRef]
- Boztug, H.; Hirschmugl, T.; Holter, W.; Lakatos, K.; Kager, L.; Trapin, D.; Pickl, W.; Forster-Waldl, E.; Boztug, K. NF-kappaB1 Haploinsufficiency Causing Immunodeficiency and EBV-Driven Lymphoproliferation. J. Clin. Immunol. 2016, 36, 533–540. [Google Scholar] [CrossRef]
- Fliegauf, M.; Bryant, V.L.; Frede, N.; Slade, C.; Woon, S.T.; Lehnert, K.; Winzer, S.; Bulashevska, A.; Scerri, T.; Leung, E.; et al. Haploinsufficiency of the NF-kappaB1 Subunit p50 in Common Variable Immunodeficiency. Am. J. Hum. Genet. 2015, 97, 389–403. [Google Scholar] [CrossRef]
- Tuijnenburg, P.; Lango Allen, H.; Burns, S.O.; Greene, D.; Jansen, M.H.; Staples, E.; Stephens, J.; Carss, K.J.; Biasci, D.; Baxendale, H.; et al. Loss-of-function nuclear factor kappaB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 2018, 142, 1285–1296. [Google Scholar] [CrossRef]
- Grossmann, M.; Metcalf, D.; Merryfull, J.; Beg, A.; Baltimore, D.; Gerondakis, S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc. Natl. Acad. Sci. USA 1999, 96, 11848–11853. [Google Scholar] [CrossRef]
- Roy, K.; Mitchell, S.; Liu, Y.; Ohta, S.; Lin, Y.S.; Metzig, M.O.; Nutt, S.L.; Hoffmann, A. A Regulatory Circuit Controlling the Dynamics of NFkappaB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity 2019, 50, 616–628. [Google Scholar] [CrossRef]
- Tarte, K.; De Vos, J.; Thykjaer, T.; Zhan, F.; Fiol, G.; Costes, V.; Reme, T.; Legouffe, E.; Rossi, J.F.; Shaughnessy, J., Jr.; et al. Generation of polyclonal plasmablasts from peripheral blood B cells: A normal counterpart of malignant plasmablasts. Blood 2002, 100, 1113–1122. [Google Scholar] [CrossRef]
- Badran, Y.R.; Dedeoglu, F.; Leyva Castillo, J.M.; Bainter, W.; Ohsumi, T.K.; Bousvaros, A.; Goldsmith, J.D.; Geha, R.S.; Chou, J. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J. Exp. Med. 2017, 214, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Sharfe, N.; Merico, D.; Karanxha, A.; Macdonald, C.; Dadi, H.; Ngan, B.; Herbrick, J.A.; Roifman, C.M. The effects of RelB deficiency on lymphocyte development and function. J. Autoimmun. 2015, 65, 90–100. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Silva, K.; Anderson, M.M.; Bhagat, G.; Klein, U. Impairment of Mature B Cell Maintenance upon Combined Deletion of the Alternative NF-kappaB Transcription Factors RELB and NF-kappaB2 in B Cells. J. Immunol. 2016, 196, 2591–2601. [Google Scholar] [CrossRef]
- Chen, K.; Coonrod, E.M.; Kumanovics, A.; Franks, Z.F.; Durtschi, J.D.; Margraf, R.L.; Wu, W.; Heikal, N.M.; Augustine, N.H.; Ridge, P.G.; et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013, 93, 812–824. [Google Scholar] [CrossRef]
- Lee, C.E.; Fulcher, D.A.; Whittle, B.; Chand, R.; Fewings, N.; Field, M.; Andrews, D.; Goodnow, C.C.; Cook, M.C. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood 2014, 124, 2964–2972. [Google Scholar] [CrossRef]
- Liu, Y.; Hanson, S.; Gurugama, P.; Jones, A.; Clark, B.; Ibrahim, M.A. Novel NFKB2 mutation in early-onset CVID. J. Clin. Immunol. 2014, 34, 686–690. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Niemela, J.E.; Sreedhara, K.; Stoddard, J.L.; Grossman, J.; Wysocki, C.A.; de la Morena, M.T.; Garofalo, M.; Inlora, J.; Snyder, M.P.; et al. Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood 2017, 130, 1553–1564. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dalla-Favera, R. Genetics of diffuse large B-cell lymphoma. Blood 2018, 131, 2307–2319. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e4. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Runge, H.F.P.; Lacy, S.; Barrans, S.; Beer, P.A.; Painter, D.; Smith, A.; Roman, E.; Burton, C.; Crouch, S.; Tooze, R.; et al. Application of the LymphGen classification tool to 928 clinically and genetically-characterised cases of diffuse large B cell lymphoma (DLBCL). Br. J. Haematol. 2021, 192, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Faumont, N.; Taoui, O.; Collares, D.; Jais, J.P.; Leroy, K.; Prevaud, L.; Jardin, F.; Molina, T.J.; Copie-Bergman, C.; Petit, B.; et al. c-Rel Is the Pivotal NF-kappaB Subunit in Germinal Center Diffuse Large B-Cell Lymphoma: A LYSA Study. Front. Oncol. 2021, 11, 638897. [Google Scholar] [CrossRef]
- Nuan-Aliman, S.; Bordereaux, D.; Thieblemont, C.; Baud, V. The Alternative RelB NF-kB Subunit Exerts a Critical Survival Function upon Metabolic Stress in Diffuse Large B-Cell Lymphoma-Derived Cells. Biomedicines 2022, 10, 348. [Google Scholar] [CrossRef]
- Lossos, I.S.; Alizadeh, A.A.; Eisen, M.B.; Chan, W.C.; Brown, P.O.; Botstein, D.; Staudt, L.M.; Levy, R. Ongoing immunoglobulin somatic mutation in germinal center B cell-like but not in activated B cell-like diffuse large cell lymphomas. Proc. Natl. Acad. Sci. USA 2000, 97, 10209–10213. [Google Scholar] [CrossRef]
- Holmes, A.B.; Corinaldesi, C.; Shen, Q.; Kumar, R.; Compagno, N.; Wang, Z.; Nitzan, M.; Grunstein, E.; Pasqualucci, L.; Dalla-Favera, R.; et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell-of-origin and outcome. JEM 2020, 217, e20200483. [Google Scholar] [CrossRef]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J. Exp. Med. 2006, 203, 311–317. [Google Scholar] [CrossRef]
- Tam, W.; Gomez, M.; Chadburn, A.; Lee, J.W.; Chan, W.C.; Knowles, D.M. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006, 107, 4090–4100. [Google Scholar] [CrossRef] [PubMed]
- Venturutti, L.; Melnick, A.M. The dangers of deja vu: Memory B cells as the cells of origin of ABC-DLBCLs. Blood 2020, 136, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Venturutti, L.; Teater, M.; Zhai, A.; Chadburn, A.; Babiker, L.; Kim, D.; Beguelin, W.; Lee, T.C.; Kim, Y.; Chin, C.R.; et al. TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate. Cell 2020, 182, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Pindzola, G.M.; Razzaghi, R.; Tavory, R.N.; Nguyen, H.T.; Morris, V.M.; Li, M.; Agarwal, S.; Huang, B.; Okada, T.; Reinhardt, H.C.; et al. Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma. Blood 2022, 140, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Suan, D.; Krautler, N.J.; Maag, J.L.V.; Butt, D.; Bourne, K.; Hermes, J.R.; Avery, D.T.; Young, C.; Statham, A.; Elliott, M.; et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 2017, 47, 1142–1153. [Google Scholar] [CrossRef]
- Young, R.M.; Wu, T.; Schmitz, R.; Dawood, M.; Xiao, W.; Phelan, J.D.; Xu, W.; Menard, L.; Meffre, E.; Chan, W.C.; et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc. Natl. Acad. Sci. USA 2015, 112, 13447–13454. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Busman-Sahay, K.; Drake, L.; Sitaram, A.; Marks, M.; Drake, J.R. Cis and trans regulatory mechanisms control AP2-mediated B cell receptor endocytosis via select tyrosine-based motifs. PLoS ONE 2013, 8, e54938. [Google Scholar] [CrossRef]
- Gross, A.J.; Lyandres, J.R.; Panigrahi, A.K.; Prak, E.T.; DeFranco, A.L. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J. Immunol. 2009, 182, 5382–5392. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Phelan, J.D.; Wright, G.W.; Haupl, B.; Huang, D.W.; Shaffer, A.L., 3rd; Young, R.M.; Wang, Z.; Zhao, H.; Yu, X.; et al. Regulation of B cell receptor-dependent NF-kappaB signaling by the tumor suppressor KLHL14. Proc. Natl. Acad. Sci. USA 2020, 117, 6092–6102. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.M.; Turer, E.E.; Liu, C.L.; Advincula, R.; Scapini, P.; Rhee, L.; Barrera, J.; Lowell, C.A.; Utz, P.J.; Malynn, B.A.; et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 2010, 33, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Vahl, J.C.; Kumar, D.; Heger, K.; Bertossi, A.; Wojtowicz, E.; Soberon, V.; Schenten, D.; Mack, B.; Reutelshofer, M.; et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 2011, 117, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Cardinez, C.; Miraghazadeh, B.; Tanita, K.; da Silva, E.; Hoshino, A.; Okada, S.; Chand, R.; Asano, T.; Tsumura, M.; Yoshida, K.; et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J. Exp. Med. 2018, 215, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Wienand, K.; Kamburov, A.; Griffin, G.K.; Chen, P.H.; Lako, A.; Redd, R.A.; et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 2019, 134, 2369–2382. [Google Scholar] [CrossRef]
- Arthur, S.E.; Jiang, A.; Grande, B.M.; Alcaide, M.; Cojocaru, R.; Rushton, C.K.; Mottok, A.; Hilton, L.K.; Lat, P.K.; Zhao, E.Y.; et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat. Commun. 2018, 9, 4001. [Google Scholar] [CrossRef]
- Nogai, H.; Wenzel, S.-S.; Hailfinger, S.; Grau, M.; Kaergel, E.; Seitz, V.; Wollert-Wulf, B.; Pfeifer, M.; Wolf, A.; Frick, M.; et al. IκB-ζ controls the constitutive NF-κB target gene network and survival of ABC DLBCL. Blood 2013, 122, 2242–2250. [Google Scholar] [CrossRef]
- Bal, E.; Kumar, R.; Hadigol, M.; Holmes, A.B.; Hilton, L.K.; Loh, J.W.; Dreval, K.; Wong, J.C.H.; Vlasevska, S.; Corinaldesi, C.; et al. Super-enhancer hypermutation alters oncogene expression in B cell lymphoma. Nature 2022, 607, 808–815. [Google Scholar] [CrossRef]
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunol. Rev. 2019, 291, 190–213. [Google Scholar] [CrossRef]
- Houldsworth, J.; Olshen, A.B.; Cattoretti, G.; Donnelly, G.B.; Teruya-Feldstein, J.; Qin, J.; Palanisamy, N.; Shen, Y.; Dyomina, K.; Petlakh, M.; et al. Relationship between REL amplification, REL function, and clinical and biologic features in diffuse large B-cell lymphomas. Blood 2004, 103, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Calado, D.P.; Wang, Z.; Frohler, S.; Kochert, K.; Qian, Y.; Koralov, S.B.; Schmidt-Supprian, M.; Sasaki, Y.; Unitt, C.; et al. An oncogenic role for alternative NF-kappaB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 2015, 11, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Calado, D.P.; Zhang, B.; Srinivasan, L.; Sasaki, Y.; Seagal, J.; Unitt, C.; Rodig, S.; Kutok, J.; Tarakhovsky, A.; Schmidt-Supprian, M.; et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 2010, 18, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Eluard, B.; Nuan-Aliman, S.; Faumont, N.; Collares, D.; Bordereaux, D.; Montagne, A.; Martins, I.; Cagnard, N.; Caly, M.; Taoui, O.; et al. The alternative RelB NF-kappaB subunit is a novel critical player in diffuse large B-cell lymphoma. Blood 2022, 139, 384–398. [Google Scholar] [CrossRef]
- Klein, U.; Heise, N. Unexpected functions of nuclear factor-kappaB during germinal center B-cell development: Implications for lymphomagenesis. Curr. Opin. Hematol. 2015, 22, 379–387. [Google Scholar] [CrossRef]
- Küppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Emmerich, F.; Theurich, S.; Hummel, M.; Haeffker, A.; Vry, M.S.; Dohner, K.; Bommert, K.; Stein, H.; Dorken, B. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J. Pathol. 2003, 201, 413–420. [Google Scholar] [CrossRef]
- Mansouri, L.; Noerenberg, D.; Young, E.; Mylonas, E.; Abdulla, M.; Frick, M.; Asmar, F.; Ljungstrom, V.; Schneider, M.; Yoshida, K.; et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood 2016, 128, 2666–2670. [Google Scholar] [CrossRef]
- Schmidt, A.; Schmitz, R.; Giefing, M.; Martin-Subero, J.I.; Gesk, S.; Vater, I.; Massow, A.; Maggio, E.; Schneider, M.; Hansmann, M.L.; et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer 2010, 49, 803–809. [Google Scholar] [CrossRef]
- Joos, S.; Menz, C.K.; Wrobel, G.; Siebert, R.; Gesk, S.; Ohl, S.; Mechtersheimer, G.; Trumper, L.; Moller, P.; Lichter, P.; et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002, 99, 1381–1387. [Google Scholar] [CrossRef]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Giefing, M.; Massow, A.; Vater, I.; Gesk, S.; Schlesner, M.; Richter, J.; Klapper, W.; Hansmann, M.L.; Siebert, R.; et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br. J. Haematol. 2012, 157, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Wlodarska, I.; Bastard, C.; Picquenot, J.M.; Hoppner, J.; Giefing, M.; Klapper, W.; Siebert, R. Chromosomal rearrangements involving the BCL3 locus are recurrent in classical Hodgkin and peripheral T-cell lymphoma. Blood 2006, 108, 401–402. [Google Scholar] [CrossRef]
- de Oliveira, K.A.; Kaergel, E.; Heinig, M.; Fontaine, J.F.; Patone, G.; Muro, E.M.; Mathas, S.; Hummel, M.; Andrade-Navarro, M.A.; Hubner, N.; et al. A roadmap of constitutive NF-kappaB activity in Hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome. Med. 2016, 8, 28. [Google Scholar] [CrossRef]
- Weniger, M.A.; Tiacci, E.; Schneider, S.; Arnolds, J.; Ruschenbaum, S.; Duppach, J.; Seifert, M.; Doring, C.; Hansmann, M.L.; Küppers, R. Human CD30+ B cells represent a unique subset related to Hodgkin lymphoma cells. J. Clin. Investig. 2018, 128, 2996–3007. [Google Scholar] [CrossRef]
- Steidl, C.; Gascoyne, R.D. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood 2011, 118, 2659–2669. [Google Scholar] [CrossRef]
- Bea, S.; Zettl, A.; Wright, G.; Salaverria, I.; Jehn, P.; Moreno, V.; Burek, C.; Ott, G.; Puig, X.; Yang, L.; et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005, 106, 3183–3190. [Google Scholar] [CrossRef]
- Weniger, M.A.; Gesk, S.; Ehrlich, S.; Martin-Subero, J.I.; Dyer, M.J.; Siebert, R.; Moller, P.; Barth, T.F. Gains of REL in primary mediastinal B-cell lymphoma coincide with nuclear accumulation of REL protein. Genes Chromosomes Cancer 2007, 46, 406–415. [Google Scholar] [CrossRef]
- Wessendorf, S.; Barth, T.F.; Viardot, A.; Mueller, A.; Kestler, H.A.; Kohlhammer, H.; Lichter, P.; Bentz, M.; Dohner, H.; Moller, P.; et al. Further delineation of chromosomal consensus regions in primary mediastinal B-cell lymphomas: An analysis of 37 tumor samples using high-resolution genomic profiling (array-CGH). Leukemia 2007, 21, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Kimm, L.R.; deLeeuw, R.J.; Savage, K.J.; Rosenwald, A.; Campo, E.; Delabie, J.; Ott, G.; Muller-Hermelink, H.K.; Jaffe, E.S.; Rimsza, L.M.; et al. Frequent occurrence of deletions in primary mediastinal B-cell lymphoma. Genes Chromosomes Cancer 2007, 46, 1090–1097. [Google Scholar] [CrossRef]
- Mottok, A.; Hung, S.S.; Chavez, E.A.; Woolcock, B.; Telenius, A.; Chong, L.C.; Meissner, B.; Nakamura, H.; Rushton, C.; Vigano, E.; et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 2019, 134, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Dunleavy, K.; Pittaluga, S.; Czuczman, M.S.; Dave, S.S.; Wright, G.; Grant, N.; Shovlin, M.; Jaffe, E.S.; Janik, J.E.; Staudt, L.M.; et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009, 113, 6069–6076. [Google Scholar] [CrossRef]
- Ruan, J.; Martin, P.; Furman, R.R.; Lee, S.M.; Cheung, K.; Vose, J.M.; Lacasce, A.; Morrison, J.; Elstrom, R.; Ely, S.; et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 690–697. [Google Scholar] [CrossRef]
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009, 114, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L., 3rd; Phelan, J.D.; Wang, J.Q.; Huang, D.; Wright, G.W.; Kasbekar, M.; Choi, J.; Young, R.M.; Webster, D.E.; Yang, Y.; et al. Overcoming Acquired Epigenetic Resistance to BTK Inhibitors. Blood Cancer Discov. 2021, 2, 630–647. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, M.; Liu, D. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 21. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef]
- Seshadri, M.R.; Melnick, A.M. Targeting MALT1 for the treatment of diffuse large B-cell lymphoma. Leuk. Lymphoma 2022, 63, 789–798. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Tuckett, A.Z.; Liou, H.C.; Doubrovina, E.; Derenzini, E.; Ouk, S.; Tsai, J.J.; Smith, O.M.; Levy, E.R.; Kreines, F.M.; et al. Characterization of a c-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-kappaB-Controlled Oxidative Stress Responses. Cancer Res. 2016, 76, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasqualucci, L.; Klein, U. NF-κB Mutations in Germinal Center B-Cell Lymphomas: Relation to NF-κB Function in Normal B Cells. Biomedicines 2022, 10, 2450. https://doi.org/10.3390/biomedicines10102450
Pasqualucci L, Klein U. NF-κB Mutations in Germinal Center B-Cell Lymphomas: Relation to NF-κB Function in Normal B Cells. Biomedicines. 2022; 10(10):2450. https://doi.org/10.3390/biomedicines10102450
Chicago/Turabian StylePasqualucci, Laura, and Ulf Klein. 2022. "NF-κB Mutations in Germinal Center B-Cell Lymphomas: Relation to NF-κB Function in Normal B Cells" Biomedicines 10, no. 10: 2450. https://doi.org/10.3390/biomedicines10102450
APA StylePasqualucci, L., & Klein, U. (2022). NF-κB Mutations in Germinal Center B-Cell Lymphomas: Relation to NF-κB Function in Normal B Cells. Biomedicines, 10(10), 2450. https://doi.org/10.3390/biomedicines10102450