

Hereditary Transthyretin-Related Amyloidosis: Genetic Heterogeneity and Early Personalized Gene Therapy
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
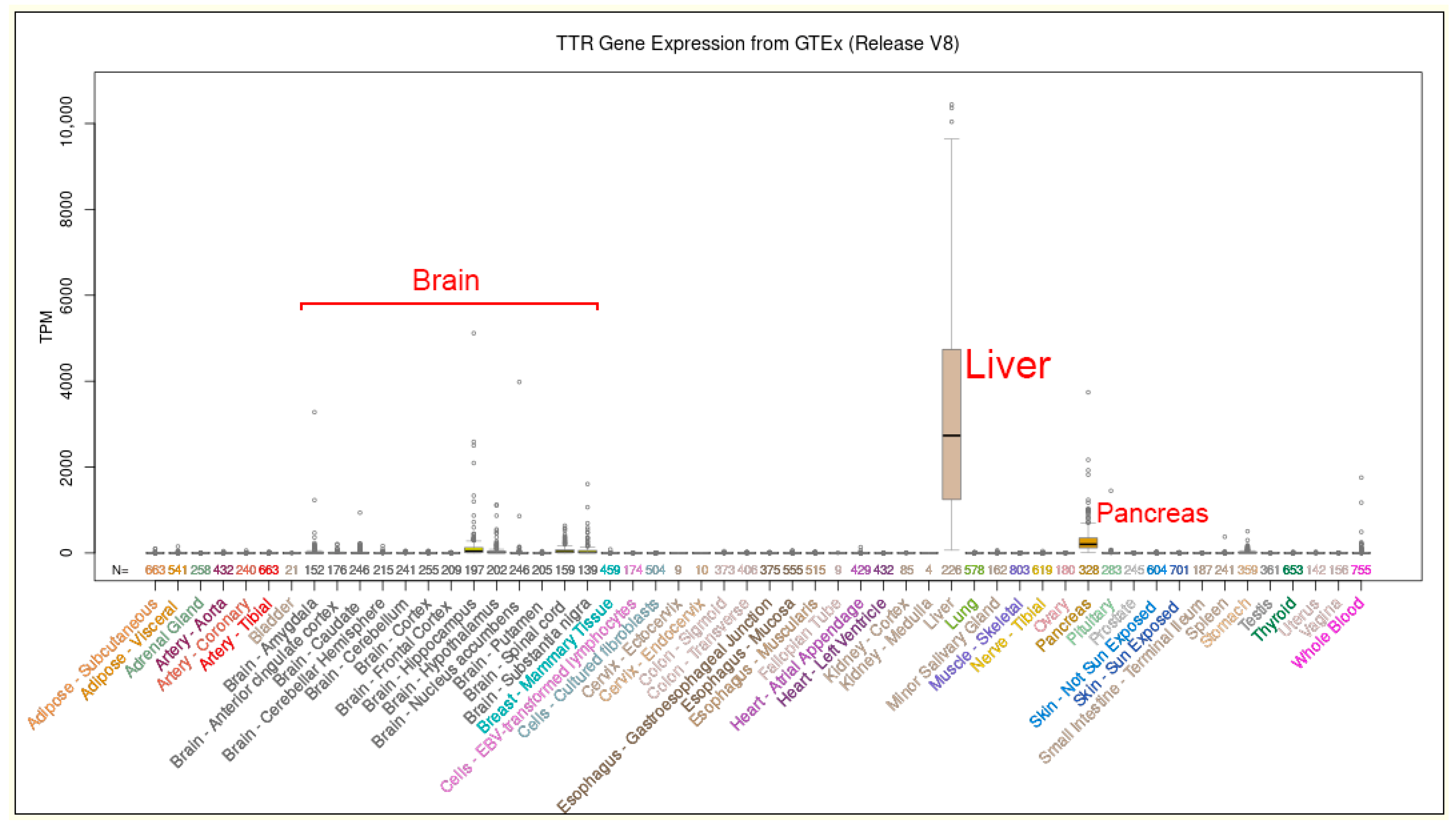{kind=link}
Abstract
1. Introduction
2. TTR Gene Variant and Hereditary Amyloidosis in Europe
- Val30Met (c.148G>A)
- Phe64Leu (c.250 T>C)
- Glu89Gln (c.325G>C)
- Thr49Ala (c.205A>G)
- Ala120Ser (c.418G>T)
- Ile68Leu (c.262A>T)
- Val122Ile (c.424G>A)
- Ala109Ser (c.385G>A)
- Thr59Lys (c.236C>A)
3. Variability and Complexity of hATTR in Sicily (Italy)
4. First Case Report of Somatic Mosaicism in hATTR Patients and Gene Conversion
5. Gene Therapy and ATTR
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andrade, C. A peculiar form of peripheral neuropathy: Familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef]
- McCutchen, S.L.; Lai, Z.; Miroy, G.J.; Kelly, J.W.; Colon, W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 1995, 34, 13527–13536. [Google Scholar] [CrossRef]
- Almeida, M.D.R.; Ferlini, A.; Forabosco, A.; Gawinowicz, M.; Costa, P.P.; Salvi, F.; Plasmati, R.; Tassinari, C.A.; Altland, K.; Saraiva, M.J. Two transthyretin Variants (TTR Ala-49 and TTR Gln-89) in two Sicilian Kindreds with Hereditary Amyloisosis. Hum. Mutat. 1992, 1, 211–215. [Google Scholar] [CrossRef]
- Lobato, L.; Beirao, I.; Guimaraes, S.M.; Droz, D.; Guimaraes, S.; Grunfeld, J.P.; Noel, L.H. Familial Amyloid Polyneuropathy Type I (Portuguese): Distribution and Characterization of Renal Amyloid Deposits. Am. J. Kidney Dis. 1998, 31, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2021, 268, 2109–2122. [Google Scholar] [CrossRef]
- Bezerra, F.; Niemietz, C.; Schmidt, H.H.J.; Zibert, A.; Guo, S.; Monia, B.P.; Gonçalves, P.; Saraiva, M.J.; Almeida, M.R. In Vitro and In Vivo Effects of SerpinA1 on the Modulation of Transthyretin Proteolysis. Int. J. Mol. Sci. 2021, 22, 9488. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11. [Google Scholar] [CrossRef]
- Si, J.-B.; Kim, B.; Kim, J.H. Transthyretin Misfolding, A Fatal Structural Pathogenesis Mechanism. Int. J. Mol. Sci. 2021, 22, 4429. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Duarte, A.; Ulloa-Aguirre, A. A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis). Int. J. Mol. Sci. 2021, 22, 13158. [Google Scholar] [CrossRef] [PubMed]
- Parman, Y.; Adams, D.; Obici, L.; Galán, L.; Guergueltcheva, V.; Suhr, O.B.; Coelho, T. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: Where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr. Opin. Neurol. 2016, 29 (Suppl. S1), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Iglseder, S.; Wanschitz, J.; Topakian, R.; Löscher, W.N.; Grisold, W. Hereditary transthyretin-related amyloidosis. Acta Neurol. Scand. 2019, 139, 92–105. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Ko, Y.H.; Runfola, M.; Rapposelli, S.; Ortore, G.; Chiellini, G.; Kim, J.H. Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis. Int. J. Mol. Sci. 2021, 22, 3488. [Google Scholar] [CrossRef]
- Federico, C.; Dugo, K.; Bruno, F.; Longo, A.M.; Grillo, A.; Saccone, S. Somatic mosaicism wih reversion to normality of a mutated transthyretin allele related to a familial amyloidotic polyneuropathy. Hum. Genet. 2017, 136, 867–873. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Mita, S.; Maeda, S.; Araki, S.; Shimada, K. Structure of the human prealbumin gene. J. Biol. Chem. 1985, 260, 12224–12227. [Google Scholar] [CrossRef]
- Nichols, W.C.; Padilla, L.M.; Benson, M.D. Prenatal Detection of a Gene for Hereditary Amyloidosis. Am. J. Med. Genet. 1989, 34, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Benson, M.D. Hereditary amyloidosis: Detection of variant prealbumin genes by restriction enzyme analysis of amplified genomic DNA sequences. Clin. Genet. 1990, 37, 44–53. [Google Scholar] [CrossRef]
- Benson, M.D.; Uemichi, T. Transthyretin Amyloidosis. Amyloid. Int. J. Exp. Clin. Investig. 1996, 3, 44–56. [Google Scholar] [CrossRef]
- Carry, B.J.; Young, K.; Fielden, S.; Kelly, M.A.; Sturm, A.C.; Avila, J.D.; Martin, C.L.; Kirchner, H.L.; Fornwalt, B.K.; Haggerty, C.M.; et al. Genomic Screening for Pathogenic Transthyretin Variants Finds Evidence of Underdiagnosed Amyloid Cardiomyopathy From Health Records. Cardio Oncol. 2021, 3, 550–561. [Google Scholar] [CrossRef]
- De Lillo, A.; Pathak, G.A.; De Angelis, F.; Di Girolamo, M.; Luigetti, M.; Sabatelli, M.; Perfetto, F.; Frusconi, S.; Manfellotto, D.; Fuciarelli, M.; et al. Epigenetic profiling of Italian patients identified methylation sites associated with hereditary transthyretin amyloidosis. Clin. Epigenetics 2020, 12, 176. [Google Scholar] [CrossRef]
- Saraiva, M.J.M.; Costa, P.P.; Almeida, M.D.R.; Banzhoff, A.; Altland, K.; Ferlini, A.; Rubboli, G.; Plasmati, R.; Tassinari, C.A.; Rome, G.; et al. Familial Amyloidotic Polyneuropathy: Transthyretin (prealbumin) variants in kindreds of Italian origin. Hum. Genet. 1988, 80, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Patrosso, M.C.; Repetto, M.; Frattini, A.; Villa, A.; Fini, S.; Salvi, F.; Vezzoni, P.; Forabosco, A. A new mutation (TTR ala-47) in the transthyretin gene associated with hereditary amyloidosis. Hum. Mutat. 1994, 4, 61–64. [Google Scholar] [CrossRef]
- Ferlini, A.; Salvi, F.; Uncini, A.; El-Chami, J.; Winter PAltland, K.; Repetto, M.; Littardi, M.; Campoleoni, A.; Vezzoni, P.; Patrosso, M.C. Homozygosity and heterozygosity for the transthyretin Leu64 mutation: Clinical, biochemical and molecular findings. Clin. Genet. 1996, 49, 10–14. [Google Scholar] [CrossRef]
- Magy, N.; Liepnieks, J.J.; Gil, H.; Kantelip, B.; Dupond, J.L.; Kluve-beckerrnan, B.; Benson, M.D. A transthyretin mutation (Tyr78Phe) associated with peripheral neuropathy, carpal tunnel syndrome and skin amyloidosis. Amyloid 2003, 10, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Jacob, E.K.; Edwards, W.D.; Zucker, M.; D’Cruz, C.; Seshan, S.V.; Crow, F.W.; Highsmith, W.E. Homozygous transthyretin mutation in an African American Male. J. Mol. Diagn. 2007, 9, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Durmuş, H.; Matur, Z.; Atmaca, M.M.; Poda, M.; Çakar, A.; Ulaş, Ü.H.; Oflazer-Serdaroğlu, P.; Deymeer, F.; Parman, Y.G. Genotypic and phenotypic presentation of transthyretin-related familial amyloid polyneuropathy (TTR-FAP) in Turkey. Neuromuscul. Disord. 2016, 26, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Benson, I.I.; Julien, J.; Liepnieks, J.; Zeldenrust, S.; Benson, M.D. A transthyretin variant (alanine 49) associated with familial amyloidotic polyneuropathy in a French family. J. Med. Genet. 1993, 30, 117–119. [Google Scholar] [CrossRef]
- Holmgren, G.; Hellman, U.; Lundgren, H.E.; Sandgren, O.; Suhr, O.B. Impact of homozygosity for an amyloidogenic transthyretin mutation on phenotype and long term outcome. J. Med. Genet. 2005, 42, 953–956. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sekijima, Y.; Ogawa, Y.; Kondo, Y.; Miyazaki, D.; Ikeda, S.I. Extremely early onset hereditary ATTR amyloidosis with G47R (p.G67R) mutation. Amyloid 2016, 23, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; Coelho, T.; Alves-Ferreira, M.; Sequeiros, J.; Mendonc, D.; Alonso, I.; Lemos, C.; Sousa, A. Familial amyloid polyneuropathy in Portugal: New genes modulating age-at-onset. Ann. Clin. Transl. Neurol. 2016, 4, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Dittloff, K.T.; Iezzi, A.; Zhong, J.X.; Mohindra, P.; Desai, T.A.; Russell, B. Transthyretin amyloid fibrils alter primary fibroblast structure, function, and inflammatory gene expression. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H149–H160. [Google Scholar] [CrossRef] [PubMed]
- Terazaki, H.; Ando, Y.; Misumi, S.; Nakamura, M.; Ando, E.; Matsunaga, N.; Shojic, S.; Okuyamaf, M.; Idetaf, H.; Nakagawa, K.; et al. A Novel Compound Heterozygote (FAP ATTR Arg104His/ATTR Val30Met) with High Serum Transthyretin (TTR) and Retinol Binding Protein (RBP) Levels. Biochem. Biophys. Res. Commun. 1999, 264, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.; Prokaeva, T.; Connors, L.H.; Falk, O.H.; Skinner, M.; Costello, C.E. Identification of a novel transthyretin Thr59Lys/Argl04His. A case of compound heterozygosity in a Chinese patient diagnosed with familial transthyretin amyloidosis. Amyloid 2002, 9, 131–140. [Google Scholar] [CrossRef]
- Iorio, A.; De Lillo, A.; De Angelis, F.; Di Girolamo, M.; Luigetti, M.; Sabatelli, M.; Pradotto, L.; Mauro, A.; Mazzeo, A.; Stancanelli, C.; et al. Non-coding variants contribute to the clinical heterogeneity of TTR amyloidosis. Eur. J. Hum. Genet. 2017, 25, 1055–1066. [Google Scholar] [CrossRef]
- Kapoor, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Clinical presentation, diagnosis and treatment of TTR amyloidosis. J. Neuromuscul. Dis. 2019, 6, 189–199. [Google Scholar] [CrossRef]
- Pelo, E.; Da Prato, L.; Ciaccheri, M.; Castelli, G.; Gori, F.; Pizzi, A.; Torricelli, F.; Marconi, G. Familial amyloid polyneuropathy with genetic anticipation associated to a gly47glu transthyretin variant in an Italian kindred. Amyloid 2002, 9, 35–41. [Google Scholar] [CrossRef]
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancanelli, C.; Gentile, L.; Baldari, S.; Carerj, S.; Toscano, A.; Vita, G. Transthyretin-related Familial Amyloid Polyneurophaty (TTRFAP): A single-center Experience in Sicily; an Italian endemic area. J. Neuromuscol. Dis. 2015, 2, S39–S48. [Google Scholar] [CrossRef]
- Kaur, D.; Tiwana, H.; Stino, A.; Sandroni, P. Autonomic neuropathies. Muscle Nerve 2020, 63, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Skare, J.C.; Saraiva, M.J.M.; Alves, I.L.; Skare, I.B.; Milunsky, A.; Cohen, A.S.; Skinner, M. A New Mutation Causing Familial Amyloidotic Polyneuropathy. Biochem. Biophys. Res. Commun. 1989, 164, 1240–1246. [Google Scholar] [CrossRef]
- Connors, L.H.; Ericsson, T.; Skare, J.; Jones, L.A.; Lewis, W.D.; Skinner, M. A simple screening test for variant transthyretins associated with familial transthyretin amyloidosis using isoelectric focusing. Biochim. Biophys. Acta 1998, 1407, 185–192. [Google Scholar] [CrossRef][Green Version]
- Sarafov, S.; Gospodinova, M.; Velina, V.G.; Kirov, A.; Teodora, C.; Todorov, T.; Todorova, A.; Tournev, I. Epidemiology of Familial Amyloid Polyneuropathy in Bulgaria. Orphanet J. Rare Dis. 2015, 10 (Suppl. S1), O2. [Google Scholar] [CrossRef]
- Gagliardi, C.; Gospodinova, M.; Longhi, S.; Milandri, A.; Cinelli, M.; Tournev, I.; Salvi, F.; Rapezzi, C. Glu89Gln transthyretin-related amyloidosis in Italy and Bulgaria: Does geographic area influence phenotype beyond the shared mutation? Orphanet J. Rare Dis. 2015, 10 (Suppl. S1), P23. [Google Scholar] [CrossRef]
- Kirov, A.; Sarafov, S.; Pavlova, Z.; Todorov, T.; Chamova, T.; Gospodinova, M.; Tournev, I.; Mitev, V.; Albena Todorova, A. Founder effect of the Glu89Gln TTR mutation in the Bulgarian population. Amyloid 2019, 26, 181–185. [Google Scholar] [CrossRef]
- Pavlova, Z.; Sarafov, S.; Todorov, T.; Kirov, A.; Chamova, T.; Gospodinova, M.; Tournev, I.; Mitev, V.; Todorova, A. Characterization of population genetic structure of hereditary transthyretin amyloidosis in Bulgaria. Amyloid 2021, 28, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F.; Scheffer, H.; Stulp, R.; Pas, H.H.; Nijenhuis, M.; Heeres, K.; Owaribe, K.; Pulkkinen, L.; Uitto, L. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene Conversion. Cell 1997, 88, 543–551. [Google Scholar] [CrossRef]
- Hirshhorn, R. In vivo reversion to normal of inherited mutations in humans. J. Med. Genet. 2003, 40, 721–728. [Google Scholar] [CrossRef]
- Chuzhanova, N.; Chen, J.M.; Bacolla, A.; Patrinos, G.P.; Férec, C.; Wells, R.D.; Cooper, D.N. Gene Conversion Causing Human Inherited Disease: Evidence for Involvement of Non-B-DNA-Forming Sequences and Recombination-Promoting Motifs in DNA Breakage and Repair. Hum. Mutat. 2009, 30, 1189–1198. [Google Scholar] [CrossRef]
- Paques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [CrossRef]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double strand break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar] [CrossRef]
- Nakamura, M.; Ando, Y.; Nagahara, S.; Sano, A.; Ochiya, T.; Maeda, S.; Kawaji, T.; Ogawa, M.; Hirata, A.; Terazaki, H. Targeted conversion of the transthyretin gene in vitro and in vivo. Gene Ther. 2004, 11, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Ando, Y. Recent advances in transthyretin amyloidosis therapy. Transl. Neurodegener. 2014, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and Consequences of Somatic Mosaicism in Humans. Nat. Rev. Genet. 2002, 3, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Trifiro, M.A. Somatic Mosaicism and Variable Expressivity. Trend Genet. 2001, 17, 79–82. [Google Scholar] [CrossRef]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef]
- Lopes, L.R.; Futema, M.; Mohammed, M.; Akhtar, M.M.; Lorenzini, M.; Pittman, A.; Syrris, P.; Elliott, P.M. Prevalence of TTR variants detected by whole-exome sequencing in hypertrophic cardiomyopathy. Amyloid 2019, 26, 243–247. [Google Scholar] [CrossRef]
- Patel, J.K.; Rosen, A.M.; Chamberlin, A.; Feldmann, B.; Antolik, C.; Zimmermann, H.; Johnston, T.; Narayana, A. Three Newly Recognized Likely Pathogenic Gene Variants Associated with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2022, 1–13. [Google Scholar] [CrossRef]
- Yordanova, I.; Pavlova, Z.; Kirov, A.; Todorov, T.; Alexiev, A.; Sarafov, S.; Mateva, L.; Chamova, T.; Gospodinova, M.; Mitev, V. Monoallelic expression of the TTR gene as a contributor to the age at onset and penetrance of TTR-related amyloidosis. Gene 2019, 705, 16–21. [Google Scholar] [CrossRef]
- Niemietz, C.; Bezerra, F.; Almeida, M.R.; Guo, S.; Monia, B.P.; Saraiva, M.J.; Schütz, P.; Schmidta, H.H.-J.; Zibert, A. SERPINA1 modulates expression of amyloidogenic transthyretin. Exp. Cell Res. 2020, 395, 112217. [Google Scholar] [CrossRef]
- Batista, A.R.; Gianni, D.; Ventosa, M.; Coelho, A.V.; Almeida, M.R.; Sena-Esteves, M.; Saraiva, M.J. Gene therapy approach to FAP: In vivo influence of T119M in TTR deposition in a transgenic V30M mouse model. Gene Ther. 2014, 21, 1041–1050. [Google Scholar] [CrossRef][Green Version]
- Bulawa, C.E.; Connelly, S.; DeVit, S.M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Brannagan, T.H.; Wang, A.K.; Coelho, T.; Waddington Cruz, M.; Polydefkis, M.J.; Dyck, P.J.; Plante-Bordeneuve, V.; Berk, J.L.; Barroso, F.; Merlini, G.; et al. Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis, a 2-year update from the open-label extension of the NEURO-TTR trial. Eur. J. Neurol. 2020, 27, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ueda, M.; Misumi, Y.; Masuda, T.; Nomura, T.; Tasaki, M.; Takamatsu, K.; Sasada, K.; Obayashi, K.; Matsui, H.; et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: Experience from a single-referral center in Japan. J. Neurol. 2017, 265, 134–140. [Google Scholar] [CrossRef]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019, 20, 2982. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, E.Y.; Oliveira, A.; Leite, J.P.; Llop, J.; Gales, L.; Quintana, J.; Cardoso, I.; Arsequell. Repurposing Benzbromarone for Familial Amyloid Polyneuropathy: A New Transthyretin Tetramer Stabilizer. Int. J. Mol. Sci. 2020, 21, 7166. [Google Scholar] [CrossRef] [PubMed]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS- the transthyretin amyloidosis outcomes survery: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 2013, 29, 63–76. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Longhi, S.; Gagliardi, C.; Milardi, A.; Manuzzi, L.; Rapezzi, C. La cardiomiopatia amiloidotica correlata alla transtiretina: Alla ricerca del trattamento eziologico. G. Ital. Di Cardiol. 2014, 15, 293–300. [Google Scholar] [CrossRef]
- Kurian, S.M.; Novais, M.; Whisenant, T.; Gelbart, T.; Buxbaum, J.N.; Kelly, J.W.; Coelho, T.; Salomon, D.R. Peripheral Blood Cell Gene Expression Diagnostic for Identifying Symptomatic Transthyretin Amyloidosis Patients: Male and Female Specific Signatures. Theranostics 2016, 6, 1792–1809. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Wixner, J.; Planté-Bordeneuve, V.; Muñoz-Beamud, F.; Lladó, L.; Gillmore, J.D.; Mazzeo, A.; Li, X.; Arum, S.; Jay, P.Y.; et al. Patisiran treatment in patients with hereditary transthyretinmediated amyloidosis with polyneuropathy after liver transplantation. Am. J. Transpl. 2022, 22, 1646–1657. [Google Scholar] [CrossRef]
- Sanguinetti, C.; Minniti, M.; Susini, V.; Caponi, L.; Panichella, G.; Castiglione, V.; Aimo, A.; Emdin, M.; Vergaro, G.; Franzini, M. The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism. Biomedicines 2022, 10, 1906. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis. In GeneReviews®; University of Washington: Seattle, DC, USA, 2021. [Google Scholar]
- Carroll, A.; Dyck, P.J.; de Carvalho, M.; Kennerson, M.; Reilly, M.M.; Kiernan, M.C.; Vucic, S. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J. Neurol. Neurosurg. Psychiatry 2022, 9, 668–678. [Google Scholar] [CrossRef]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Plante-Bordeneuve, V.; Schmidt, H.H.J.; Merlini, G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J. Am. Coll. Cardiol. 2015, 66, 2451–2466. [Google Scholar] [CrossRef]
- Steinhoff, J.S.; Lass, A.; Schupp, M. Biological Functions of RBP4 and Its Relevance for Human Diseases. Front. Physiol. 2021, 12, 659977. [Google Scholar] [CrossRef]
- Shestak, A.G.; Bukaeva, A.A.; Saber, S.; Zaklyazminskaya, E.V. Allelic Dropout Is a Common Phenomenon That Reduces the Diagnostic Yield of PCR-Based Sequencing of Targeted Gene Panels. Front. Genet. 2021, 12, 620337. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugo, K.; Bruno, F.; Sturiale, V.; Brancato, D.; Saccone, S.; Federico, C. Hereditary Transthyretin-Related Amyloidosis: Genetic Heterogeneity and Early Personalized Gene Therapy. Biomedicines 2022, 10, 2394. https://doi.org/10.3390/biomedicines10102394
Dugo K, Bruno F, Sturiale V, Brancato D, Saccone S, Federico C. Hereditary Transthyretin-Related Amyloidosis: Genetic Heterogeneity and Early Personalized Gene Therapy. Biomedicines. 2022; 10(10):2394. https://doi.org/10.3390/biomedicines10102394
Chicago/Turabian StyleDugo, Ketty, Francesca Bruno, Valentina Sturiale, Desiree Brancato, Salvatore Saccone, and Concetta Federico. 2022. "Hereditary Transthyretin-Related Amyloidosis: Genetic Heterogeneity and Early Personalized Gene Therapy" Biomedicines 10, no. 10: 2394. https://doi.org/10.3390/biomedicines10102394
APA StyleDugo, K., Bruno, F., Sturiale, V., Brancato, D., Saccone, S., & Federico, C. (2022). Hereditary Transthyretin-Related Amyloidosis: Genetic Heterogeneity and Early Personalized Gene Therapy. Biomedicines, 10(10), 2394. https://doi.org/10.3390/biomedicines10102394