
Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research

 ,
,  ,
,
Abstract
:1. Introduction
2. The Human Plasma Proteome and Its Complexity
3. Cardiovascular Diseases and Related Biomarkers
4. Recent Developments in Assaying the Human Plasma Proteome by Proteomics Methods
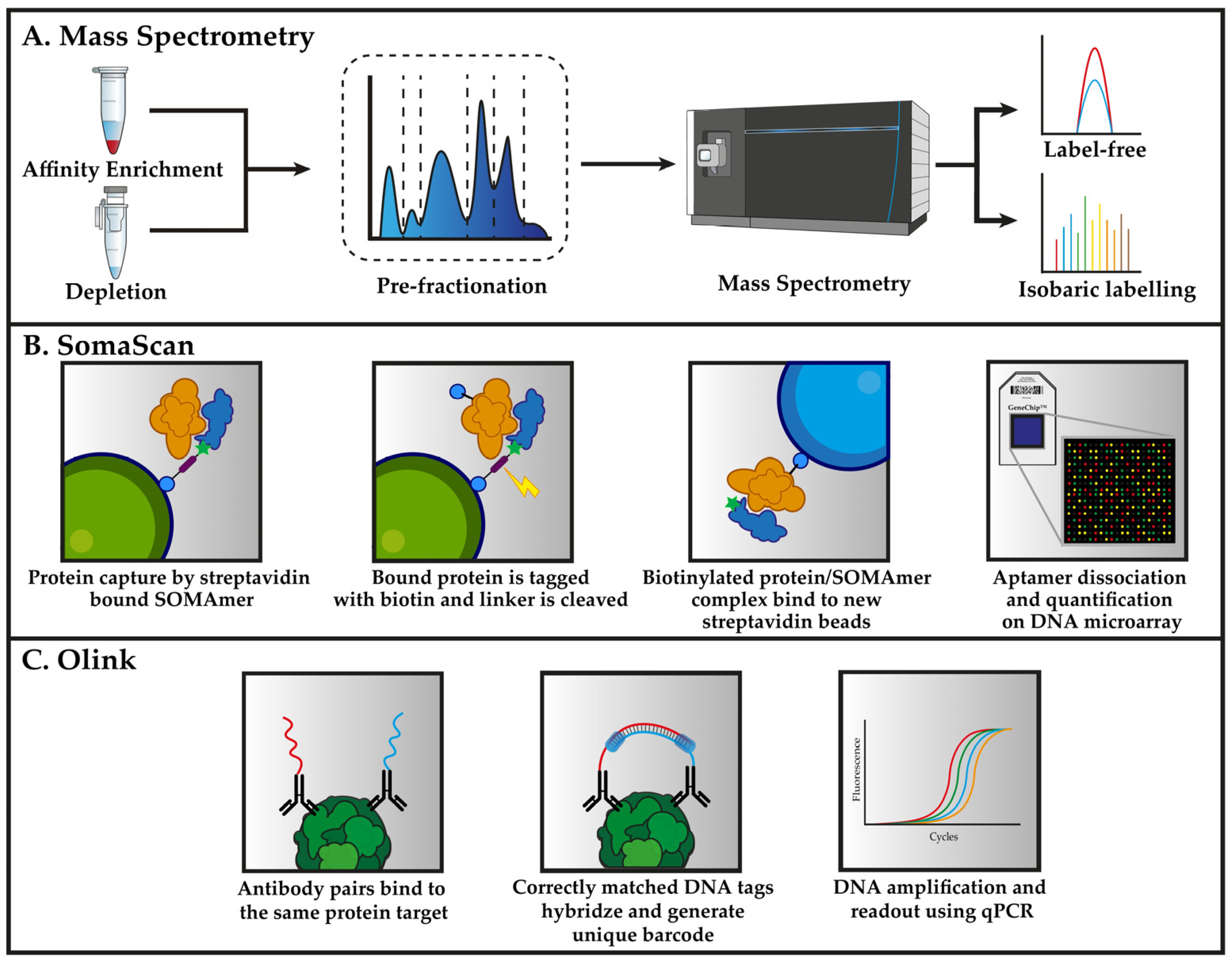
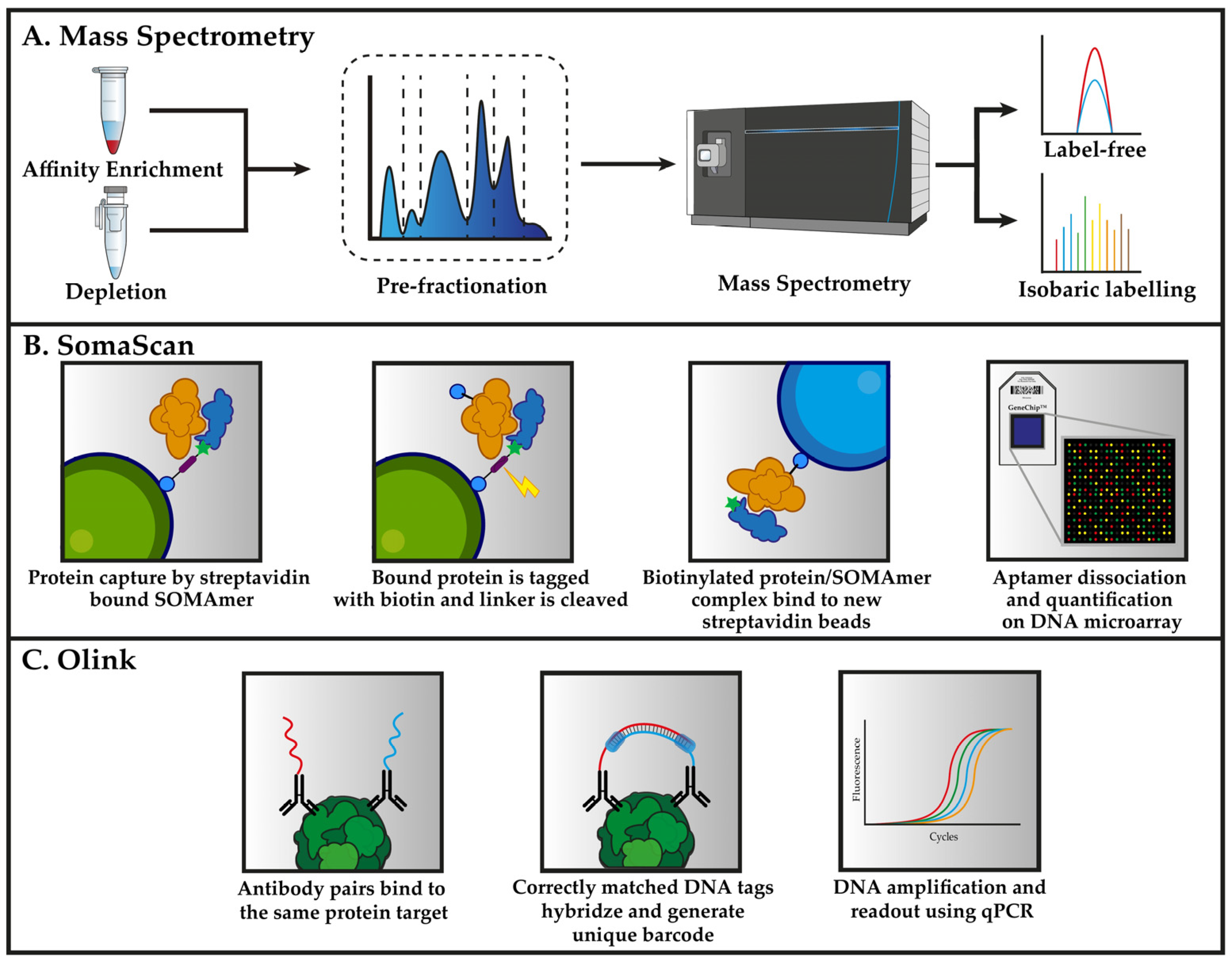
4.1. Mass Spectrometry-Based Plasma Proteomics
4.1.1. Preanalytical Steps in Mass Spectrometry-Based Plasma Proteomics
4.1.2. MS-Based Proteomics: LC-MSMS Analysis
4.1.3. Quantitative MS-Based Proteomics
4.2. Affinity-Based Proteomics Methods
4.2.1. Aptamer Microarrays (SomaScan)
4.2.2. Proximity Extension Assays (Olink)
4.2.3. Microbead-Based Multiplex Immunoassay (xMAP)
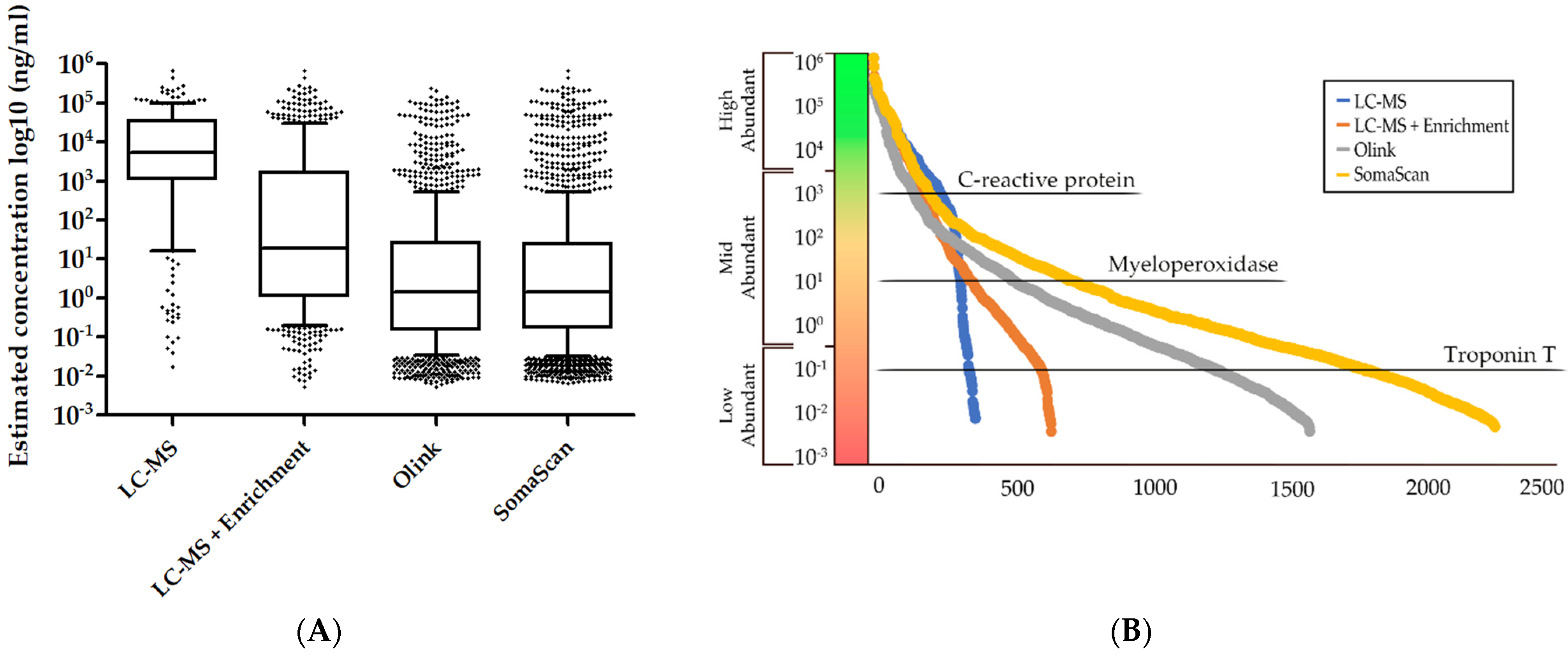
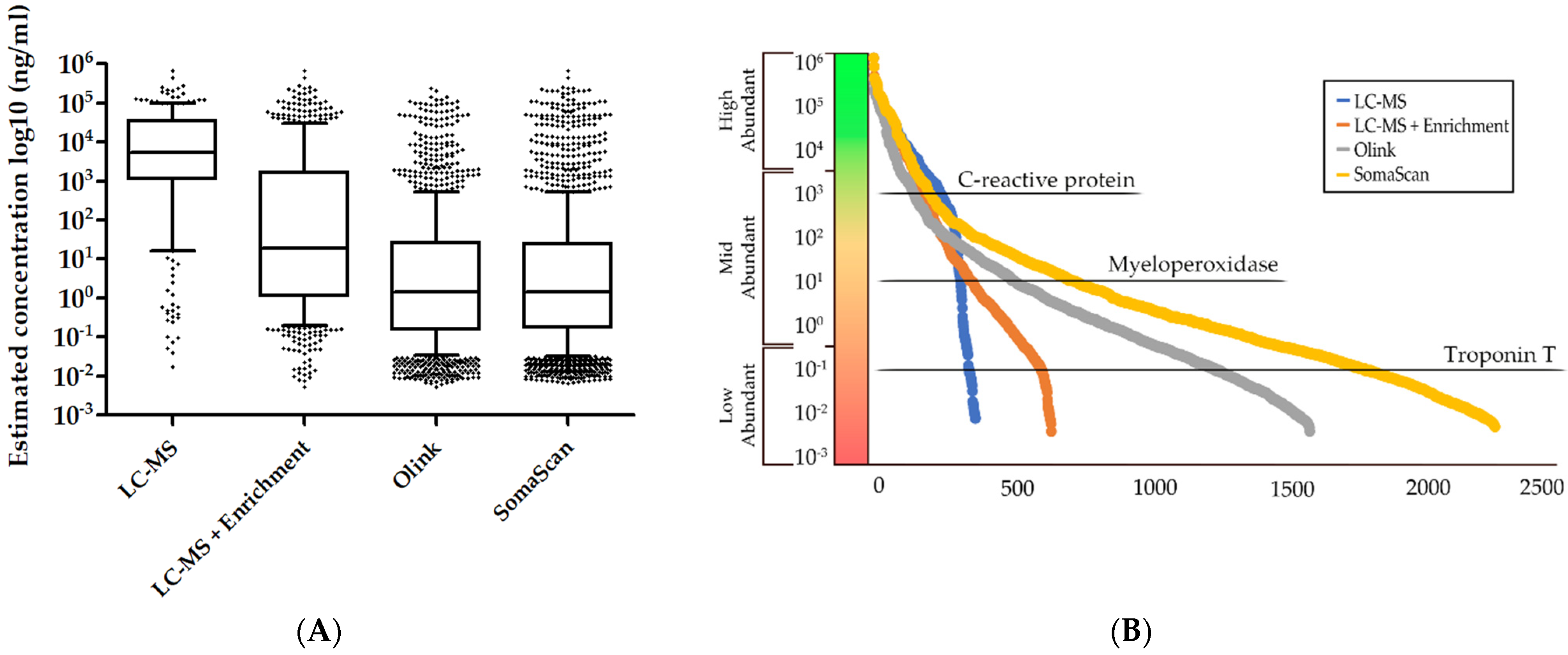
5. Strengths and Limitations of the Current, Major Plasma Proteomics Technologies
6. Recent Developments in Plasma Proteomics of Cardiovascular Diseases
7. Conclusive Remarks and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2020, 395, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, R.; Vasan, R.S. Biomarkers in cardiovascular disease: Statistical assessment and section on key novel heart failure biomarkers. Trends Cardiovasc. Med. 2017, 27, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Beach, S.R.H.; Simons, R.L.; Lendasse, A.; Penaluna, B.; Philibert, R.A. Blood-Based Biomarkers for Predicting the Risk for Five-Year Incident Coronary Heart Disease in the Framingham Heart Study via Machine Learning. Genes 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedl, T.J.; Gil, R.S.; Cheng, F.; Rayner, S.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef] [Green Version]
- Khalilpour, A.; Kilic, T.; Alvarez, M.M.; Yazdi, I.K. Proteomic-based biomarker discovery for development of next generation diagnostics. Appl. Microbiol. Biotechnol. 2017, 101, 475–491. [Google Scholar] [CrossRef]
- Uhlén, M.; Karlsson, M.J.; Hober, A.; Svensson, A.-S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal. 2019, 12, eaaz0274. [Google Scholar] [CrossRef] [Green Version]
- Farrah, T.; Deutsch, E.; Omenn, G.S.; Campbell, D.S.; Sun, Z.; Bletz, J.A.; Mallick, P.; Katz, J.E.; Malmström, J.; Ossola, R.; et al. A High-Confidence Human Plasma Proteome Reference Set with Estimated Concentrations in PeptideAtlas. Mol. Cell. Proteom. 2011, 10, M110.006353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Poljak, A.; Ali, S.A.; Zhong, L.; Raftery, M.J.; Sachdev, P. Extending the Depth of Human Plasma Proteome Coverage Using Simple Fractionation Techniques. J. Proteome Res. 2021, 20, 1261–1279. [Google Scholar] [CrossRef]
- Suski, M.; Bokiniec, R.; Szwarc-Duma, M.; Madej, J.; Bujak-Gizycka, B.; Kwinta, P.; Borszewska-Kornacka, M.K.; Revhaug, C.; Baumbusch, L.O.; Saugstad, O.D.; et al. Prospective plasma proteome changes in preterm infants with different gestational ages. Pediatr. Res. 2018, 84, 104–111. [Google Scholar] [CrossRef]
- Anderson, N.L. The Human Plasma Proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Daghri, N.M.; Torretta, E.; Capitanio, D.; Fania, C.; Guerini, F.R.; Sabico, S.B.; Clerici, M.; Gelfi, C. Intermediate and low abundant protein analysis of vitamin D deficient obese and non-obese subjects by MALDI-profiling. Sci. Rep. 2017, 7, 12633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.; Kopylov, A.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, M.J.; Forbes, A.J.; Boyne, M.T.; Kim, Y.-B.; Robinson, D.E.; Kelleher, N.L. Precise and Parallel Characterization of Coding Polymorphisms, Alternative Splicing, and Modifications in Human Proteins by Mass Spectrometry. Mol. Cell. Proteom. 2005, 4, 1002–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddy, A.K.; White, M.Y.; Cordwell, S.J. Functional decorations: Post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahn, M.; Michel, P.-A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; de Laval, V.R.; et al. The neXtProt knowledgebase in 2020: Data, tools and usability improvements. Nucleic Acids Res. 2019, 48, D328–D334. [Google Scholar] [CrossRef] [Green Version]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, E.C.; Cravatt, B.F.; Fenselau, C.; Garcia, A.B.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archakov, A.; Zgoda, V.; Kopylov, A.; Naryzhny, S.; Chernobrovkin, A.; Ponomarenko, E.; Lisitsa, A. Chromosome-centric approach to overcoming bottlenecks in the Human Proteome Project. Expert Rev. Proteom. 2012, 9, 667–676. [Google Scholar] [CrossRef]
- Mokou, M.; Lygirou, V.; Vlahou, A.; Mischak, H. Proteomics in cardiovascular disease: Recent progress and clinical implication and implementation. Expert Rev. Proteom. 2017, 14, 117–136. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, N.; Incollingo, P.; Tammaro, V.; Peluso, G.; Rupealta, N.; Chiacchio, G.; Sandoval Sotelo, M.L.; Minieri, G.; Pisani, A.; Riccio, E.; et al. Diagnostic, Predictive, Prognostic, and Therapeutic Molecular Biomarkers in Third Millennium: A Breakthrough in Gastric Cancer. BioMed Res. Int. 2017, 2017, 7869802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sechidis, K.; Papangelou, K.; Metcalfe, P.D.; Svensson, D.; Weatherall, J.; Brown, G. Distinguishing prognostic and predictive biomarkers: An information theoretic approach. Bioinformatics 2018, 34, 3365–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res. 2017, 16, 4299–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, G.; List, M.; Zhang, J.D. Tissue heterogeneity is prevalent in gene expression studies. NAR Genom. Bioinform. 2021, 3, lqab077. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.L.; Duffy, S.; Lang, S.H. Impact of tumor heterogeneity and tissue sampling for genetic mutation testing: A systematic review and post hoc analysis. J. Clin. Epidemiol. 2020, 126, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Pernemalm, M.; Sandberg, A.; Zhu, Y.; Boekel, J.; Tamburro, D.; Schwenk, J.M.; Björk, A.; Wahren-Herlenius, M.; Åmark, H.; Östenson, C.-G.; et al. In-depth human plasma proteome analysis captures tissue proteins and transfer of protein variants across the placenta. eLife 2019, 8, e41608. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, L.J.; Li, M.; Yarbrough, W.G.; Slebos, R.J.C.; Liebler, D.C. Global Stability of Plasma Proteomes for Mass Spectrometry-Based Analyses. Mol. Cell. Proteom. 2012, 11, M111.014340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giavarina, D.; Lippi, G. Blood venous sample collection: Recommendations overview and a checklist to improve quality. Clin. Biochem. 2017, 50, 568–573. [Google Scholar] [CrossRef]
- Grankvist, K.; Gomez, R.; Nybo, M.; Lima-Oliveira, G.; von Meyer, A. Preanalytical aspects on short- and long-term storage of serum and plasma. Diagnosis 2019, 6, 51–56. [Google Scholar] [CrossRef]
- Simundic, A.-M.; Church, S.; Cornes, M.P.; Grankvist, K.; Lippi, G.; Nybo, M.; Nikolac, N.; Van Dongen-Lases, E.; Eker, P.; Kovalevskaya, S.; et al. Compliance of blood sampling procedures with the CLSI H3-A6 guidelines: An observational study by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) working group for the preanalytical phase (WG-PRE). Clin. Chem. Lab. Med. CCLM 2014, 53, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Danese, E.; Montagnana, M. An historical approach to the diagnostic biomarkers of acute coronary syndrome. Ann. Transl. Med. 2016, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Babuin, L. Troponin: The biomarker of choice for the detection of cardiac injury. Can. Med Assoc. J. 2005, 173, 1191–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Jia, Y.; Zhu, B. BNP and NT-proBNP as Diagnostic Biomarkers for Cardiac Dysfunction in Both Clinical and Forensic Medicine. Int. J. Mol. Sci. 2019, 20, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostovenko, E.; Scott, H.C.; Klychnikov, O.; Dalebout, H.; Deelder, A.M.; Palmblad, M. Protein Fractionation for Quantitative Plasma Proteomics by Semi-Selective Precipitation. J. Proteom. Bioinform. 2012, 5, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Boschetti, E.; Righetti, P.G. The ProteoMiner in the proteomic arena: A non-depleting tool for discovering low-abundance species. J. Proteom. 2008, 71, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhou, J.-Y.; Gritsenko, M.A.; Hossain, M.; Camp, D.G.; Smith, R.D.; Qian, W.-J. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography–mass spectrometry for human plasma proteomics biomarker discovery. Methods 2012, 56, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, L.A.; Ky, B.; Barnhart, K.T.; Speicher, D.W. In-Depth, Reproducible Analysis of Human Plasma Using IgY 14 and SuperMix Immunodepletion. Methods Mol. Biol. 2017, 1619, 81–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palstrøm, N.B.; Rasmussen, L.M.; Beck, H.C. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. Int. J. Mol. Sci. 2020, 21, 5903. [Google Scholar] [CrossRef]
- Beck, H.C.; Jensen, L.O.; Gils, C.; Ilondo, A.M.M.; Frydland, M.; Hassager, C.; Møller-Helgestad, O.K.; Møller, J.E.; Rasmussen, L.M. Proteomic Discovery and Validation of the Confounding Effect of Heparin Administration on the Analysis of Candidate Cardiovascular Biomarkers. Clin. Chem. 2018, 64, 1474–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debrabant, B.; Halekoh, U.; Soerensen, M.; Møller, J.E.; Hassager, C.; Frydland, M.; Palstrøm, N.; Hjelmborg, J.; Beck, H.C.; Rasmussen, L.M. STEMI, Cardiogenic Shock, and Mortality in Patients Admitted for Acute Angiography: Associations and Predictions from Plasma Proteome Data. Shock 2021, 55, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.O.; Sui, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81. [Google Scholar] [CrossRef] [PubMed]
- DuPree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.G.; Sokolowska, I.; Wetie, A.G.N.; Channaveerappa, D.; Dupree, E.J.; Jayathirtha, M.; Aslebagh, R.; Wormwood, K.L.; Darie, C.C. Mass Spectrometry for Proteomics-Based Investigation. Adv. Exp. Med. Biol. 2019, 1140, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef]
- Baker, E.S.; Liu, T.; Petyuk, A.V.; Burnum-Johnson, E.K.; Ibrahim, Y.M.; Anderson, A.G.; Smith, R.D. Mass spectrometry for translational proteomics: Progress and clinical implications. Genome Med. 2012, 4, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tucholska, M.; Knight, J.D.; Lambert, J.-P.; Tate, S.; Larsen, B.; Gingras, A.-C.; Bandeira, N. MSPLIT-DIA: Sensitive peptide identification for data-independent acquisition. Nat. Methods 2015, 12, 1106–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilaire, P.B.S.; Rousseau, K.; Seyer, A.; Dechaumet, S.; Damont, A.; Junot, C.; Fenaille, F. Comparative Evaluation of Data Dependent and Data Independent Acquisition Workflows Implemented on an Orbitrap Fusion for Untargeted Metabolomics. Metabolites 2020, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef]
- Orsburn, B. Proteome Discoverer—A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Röst, H.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkovits, K.; Pacharra, S.; Pfeiffer, K.; Steinbach, S.; Eisenacher, M.; Marcus, K.; Uszkoreit, J. Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-based Data-independent Acquisition. Mol. Cell. Proteom. 2020, 19, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberger, G.; Liu, Y.; Röst, H.L.; Ludwig, C.; Buil, A.; Bensimon, A.; Soste, M.; Spector, T.D.; Dermitzakis, E.T.; Collins, B.C.; et al. Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat. Biotechnol. 2017, 35, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, A.; Damoc, E.; Hauschild, J.-P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2011, 10, M111.011015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical Analysis of Membrane Proteome Expression Changes in Saccharomyces cerevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, C.; Hansen, R.; Baumann, S.; Kublik, A.; Nielsen, P.H.; Adrian, L.; von Bergen, M.; Jehmlich, N.; Seifert, J. Comparison of targeted peptide quantification assays for reductive dehalogenases by selective reaction monitoring (SRM) and precursor reaction monitoring (PRM). Anal. Bioanal. Chem. 2014, 406, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Ronsein, G.E.; Pamir, N.; von Haller, P.D.; Kim, D.S.; Oda, M.N.; Jarvik, G.P.; Vaisar, T.; Heinecke, J.W. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J. Proteom. 2015, 113, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.P.; Larsen, M.R.; Mickley, H.; Saaby, L.; Diederichsen, A.C.; Lambrechtsen, J.; Rasmussen, L.M.; Overgaard, M. Plasma proteome profiling of atherosclerotic disease manifestations reveals elevated levels of the cytoskeletal protein vinculin. J. Proteom. 2014, 101, 141–153. [Google Scholar] [CrossRef]
- Lindemann, C.; Thomanek, N.; Hundt, F.; Lerari, T.; Meyer, H.E.; Wolters, D.; Marcus, K. Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol. Chem. 2017, 398, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.G. Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods Protoc. 2019, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prianichnikov, N.; Koch, H.; Koch, S.; Lubeck, M.; Heilig, R.; Brehmer, S.; Fischer, R.; Cox, J. MaxQuant Software for Ion Mobility Enhanced Shotgun Proteomics. Mol. Cell. Proteom. 2020, 19, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauniyar, N.; Yates, J.R. Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [Green Version]
- Unwin, R.; Pierce, A.; Watson, R.B.; Sternberg, D.W.; Whetton, A.D. Quantitative Proteomic Analysis Using Isobaric Protein Tags Enables Rapid Comparison of Changes in Transcript and Protein Levels in Transformed Cells. Mol. Cell. Proteom. 2005, 4, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, A.T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef]
- Leitner, A. A review of the role of chemical modification methods in contemporary mass spectrometry-based proteomics research. Anal. Chim. Acta 2018, 1000, 2–19. [Google Scholar] [CrossRef]
- Hogrebe, A.; Von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Sonnett, M.; Yeung, E.; Wühr, M. Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Anal. Chem. 2018, 90, 5032–5039. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciampa, E.; Li, Y.; Dillon, S.; Lecarpentier, E.; Sorabella, L.; Libermann, T.A.; Karumanchi, S.A.; Hess, P.E. Cerebrospinal Fluid Protein Changes in Preeclampsia. Hypertension 2018, 72, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.; Libermann, T.; Bowditch, H.; Das, G.; Diakos, N.; Huggins, G.; Rastegar, H.; Chen, F.; Rowin, E.; Maron, M.; et al. Plasma Proteomic Profiling in Hypertrophic Cardiomyopathy Patients before and after Surgical Myectomy Reveals Post-Procedural Reduction in Systemic Inflammation. Int. J. Mol. Sci. 2021, 22, 2474. [Google Scholar] [CrossRef]
- Billing, A.M.; Ben Hamidane, H.; Bhagwat, A.M.; Cotton, R.J.; Dib, S.S.; Kumar, P.; Hayat, S.; Goswami, N.; Suhre, K.; Rafii, A.; et al. Complementarity of SOMAscan to LC-MS/MS and RNA-seq for quantitative profiling of human embryonic and mesenchymal stem cells. J. Proteom. 2017, 150, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Welton, J.L.; Brennan, P.; Gurney, M.; Webber, J.P.; Spary, L.; Gil Carton, D.; Falcón-Pérez, J.M.; Walton, S.P.; Mason, M.D.; Tabi, Z.; et al. Proteomics analysis of vesicles isolated from plasma and urine of prostate cancer patients using a multiplex, aptamer-based protein array. J. Extracell. Vesicles 2016, 5, 31209. [Google Scholar] [CrossRef] [Green Version]
- Chirinos, J.A.; Cohen, J.B.; Zhao, L.; Hanff, T.; Sweitzer, N.; Fang, J.; Corrales-Medina, V.; Ammar, R.; Morley, M.; Zamani, P.; et al. Clinical and Proteomic Correlates of Plasma ACE2 (Angiotensin-Converting Enzyme 2) in Human Heart Failure. Hypertension 2020, 76, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.-H.; Prabath, J.-F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing Candidates of Post–Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020, 142, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Raafs, A.; Verdonschot, J.; Ferreira, J.P.; Wang, P.; Collier, T.; Henkens, M.; Björkman, J.; Boccanelli, A.; Clark, A.L.; Delles, C.; et al. Identification of sex-specific biomarkers predicting new-onset heart failure. ESC Hear. Fail. 2021, 8, 3512–3520. [Google Scholar] [CrossRef]
- Wallentin, L.; Eriksson, N.; Olszowka, M.; Grammer, T.B.; Hagström, E.; Held, C.; Kleber, M.E.; Koenig, W.; März, W.; Stewart, R.A.H.; et al. Plasma proteins associated with cardiovascular death in patients with chronic coronary heart disease: A retrospective study. PLoS Med. 2021, 18, e1003513. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.S.; Paniagua, S.M.; Guseh, J.; Bhambhani, V.; Zanni, M.V.; Courchesne, P.; Lyass, A.; Larson, M.G.; Levy, D.; Ho, J.E. Sex Differences in Circulating Biomarkers of Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 74, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, N.; Xiang, Y.; Wu, L.; Li, C.; Yuan, Z.; Jia, X.; Zhang, Z.; Zhong, L.; Li, Y. Prognostic value of chemokines in patients with newly diagnosed atrial fibrillation. Int. J. Cardiol. 2020, 320, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Skalnikova, H.K.; Cizkova, J.; Cervenka, J.; Vodicka, P. Advances in Proteomic Techniques for Cytokine Analysis: Focus on Melanoma Research. Int. J. Mol. Sci. 2017, 18, 2697. [Google Scholar] [CrossRef] [Green Version]
- Kiouptsi, K.; Pontarollo, G.; Todorov, H.; Braun, J.; Jäckel, S.; Koeck, T.; Bayer, F.; Karwot, C.; Karpi, A.; Gerber, S.; et al. Germ-free housing conditions do not affect aortic root and aortic arch lesion size of late atherosclerotic low-density lipoprotein receptor-deficient mice. Gut Microbes 2020, 11, 1809–1823. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth Differentiation Factor 11 Is a Circulating Factor that Reverses Age-Related Cardiac Hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olink Explore 3072 Complete Assay List. Available online: https://www.olink.com/resources-support/document-download-center/ (accessed on 30 November 2021).
- Williams, S.A.; Kivimaki, M.; Langenberg, C.; Hingorani, A.D.; Casas, J.P.; Bouchard, C.; Jonasson, C.; Sarzynski, M.A.; Shipley, M.J.; Alexander, L.; et al. Plasma protein patterns as comprehensive indicators of health. Nat. Med. 2019, 25, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, J.; Si, T. Recent advances in high-throughput mass spectrometry that accelerates enzyme engineering for biofuel research. BMC Energy 2020, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, J.-P.; Peterson, A.C.; Couzijn, E.; Denisov, E.; Chernyshev, D.; Hock, C.; Stewart, H.; Hartmer, R.; Grinfeld, D.; Thoeing, C.; et al. A Novel Family of Quadrupole-Orbitrap Mass Spectrometers for a Broad Range of Analytical Applications. Preprints 2020, 1, 2020060111. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, J.R.; Wybenga-Groot, L.E.; Tong, J.; Bache, N.; Tsao, M.; Moran, M.F. Evosep One Enables Robust Deep Proteome Coverage Using Tandem Mass Tags while Significantly Reducing Instrument Time. J. Proteome Res. 2019, 18, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.-S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e4. [Google Scholar] [CrossRef]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Raffield, L.M.; Dang, H.; Pratte, K.A.; Jacobson, S.; Gillenwater, L.A.; Ampleford, E.; Barjaktarevic, I.; Basta, P.; Clish, C.B.; Comellas, A.P.; et al. Comparison of Proteomic Assessment Methods in Multiple Cohort Studies. Proteomics 2020, 20, e1900278. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Tworoger, S.S.; Stampfer, M.J.; Dillon, S.T.; Gu, X.; Sawyer, S.J.; Chan, A.T.; Libermann, T.A.; Eliassen, A.H. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci. Rep. 2018, 8, 8382. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Koul, S.; Egerstedt, A.; Smith, J.G.; Noc, M.; Lang, I.; Holzer, M.; Clemmensen, P.; Gidlöf, O.; Metzler, B.; et al. Using proximity extension proteomics assay to identify biomarkers associated with infarct size and ejection fraction after ST-elevation myocardial infarction. Sci. Rep. 2020, 10, 18663. [Google Scholar] [CrossRef]
- Kulasingam, A.; Hvas, A.-M.; Grove, E.L.; Funck, K.L.; Kristensen, S.D. Detection of biomarkers using a novel proximity extension assay in patients with ST-elevation myocardial infarction. Thromb. Res. 2018, 172, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Näsström, B.; Olivecrona, G.; Olivecrona, T.; Stegmayr, B. Lipoprotein lipase during continuous heparin infusion: Tissue stores become partially depleted. J. Lab. Clin. Med. 2001, 138, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Sidloff, D.; Stather, P.; Dattani, N.; Bown, M.; Thompson, J.; Sayers, R.; Choke, E. Aneurysm Global Epidemiology Study: Public health measures can further reduce abdominal aortic aneurysm mortality. Circulation 2014, 129, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Henriksson, A.E.; Lindqvist, M.; Sihlbom, C.; Bergström, J.; Bylund, D. Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantita tive Proteomics. Proteomes 2018, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Molina-Sánchez, P.; Jorge, I.; Martinez-Pinna, R.; Blanco-Colio, L.M.; Tarin, C.; Torres-Fonseca, M.M.; Esteban, M.; Laustsen, J.; Ramos-Mozo, P.; Calvo, E.; et al. ApoA-I/HDL-C levels are inversely associated with abdominal aortic aneurysm progression. Thromb. Haemost. 2015, 113, 1335–1346. [Google Scholar] [CrossRef]
- Nana, P.; Dakis, K.; Brodis, A.; Spanos, K.; Kouvelos, G. Circulating Biomarkers for the Prediction of Abdominal Aortic Aneurysm Growth. J. Clin. Med. 2021, 10, 1718. [Google Scholar] [CrossRef]
- Ngo, D.; Sinha, S.; Shen, D.; Kuhn, E.W.; Keyes, M.J.; Shi, X.; Benson, M.D.; O’Sullivan, J.; Keshishian, H.; Farrell, L.A.; et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation 2016, 134, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, J.; Ngo, D.; Finkel, N.; Pitts, R.; Gleim, S.; Benson, M.D.; Keyes, M.J.; Farrell, L.A.; Morgan, T.; Jennings, L.L.; et al. Application of Large-Scale Aptamer-Based Proteomic Profiling to Planned Myocardial Infarctions. Circulation 2018, 137, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Yang, Q.; Ngo, D.; Zhu, Y.; Shen, D.; Farrell, L.A.; Sinha, S.; Keyes, M.J.; Vasan, R.S.; Larson, M.G.; et al. Genetic Architecture of the Cardiovascular Risk Proteome. Circulation 2018, 137, 1158–1172. [Google Scholar] [CrossRef]
- Gui, H.; She, R.; Luzum, J.; Li, J.; Bryson, T.D.; Pinto, Y.; Sabbah, H.N.; Williams, L.K.; Lanfear, D.E. Plasma Proteomic Profile Predicts Survival in Heart Failure with Reduced Ejection Fraction. Circ. Genom. Precis. Med. 2021, 14, 003140. [Google Scholar] [CrossRef]
- Staerk, L.; Preis, S.R.; Lin, H.; Lubitz, S.A.; Ellinor, P.T.; Levy, D.; Benjamin, E.J.; Trinquart, L. Protein Biomarkers and Risk of Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2020, 13, e007607. [Google Scholar] [CrossRef]
- Ku, E.J.; Cho, K.-C.; Lim, C.; Kang, J.W.; Oh, J.W.; Choi, Y.R.; Park, J.-M.; Han, N.-Y.; Oh, J.J.; Oh, T.J.; et al. Discovery of plasma biomarkers for predicting the severity of coronary artery atherosclerosis by quantitative proteomics. BMJ Open Diabetes Res. Care 2020, 8, e001152. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Common Errors Related to Venous Blood Collection |
|---|
| Mislabeling |
| Wrong collection tube |
| Insufficient volume drawn |
| Sample contamination |
| Incorrect sample handling |
| Hemolysis |
| Prolonged storage at incorrect temperature |
| MS-Based | Aptamer-Based | Immunoaffinity-Based | |
|---|---|---|---|
| Types | Discovery (DDA) Targeted (DIA, SRM/MRM) | Nucleic acid-binders (aptamers) | Antibody-dependent |
| Quantification | Relative: Label-free, isobaric labelling Absolute: targeted stable isotope standards | Relative only | Relative (Olink) or absolute |
| Proteome coverage in plasma | Method/instrument dependent, 250 to 2000 | ≥7000 | Depends on the kits used, 1 to 384 (Olink) |
| Sample throughput | Protocol-dependent, high throughput is achievable | High throughput | High throughput |
| Reproducibility | Low intra-assay CV with isobaric labelling or targeted. Generally, higher with label free | Low intra-assay CV | Low intra-assay CV |
| Specificity | High | Modest to high, known issues with unspecific binding and cross-reactivity | Modest to high when using antibody pairs (Olink) |
| Identification of different proteoforms (PTMs, isoforms, etc.) | Easy. Existing workflows available | Challenging. Requires development of specific aptamers | Challenging. Requires specific antibodies |
| Availability/Expertise required | Mostly in-house, which requires high level of expertise. Commercial solutions exist. | Commercial—no expertise required | Commercial or in-house |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palstrøm, N.B.; Matthiesen, R.; Rasmussen, L.M.; Beck, H.C. Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research. Biomedicines 2022, 10, 162. https://doi.org/10.3390/biomedicines10010162
Palstrøm NB, Matthiesen R, Rasmussen LM, Beck HC. Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research. Biomedicines. 2022; 10(1):162. https://doi.org/10.3390/biomedicines10010162
Chicago/Turabian StylePalstrøm, Nicolai Bjødstrup, Rune Matthiesen, Lars Melholt Rasmussen, and Hans Christian Beck. 2022. "Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research" Biomedicines 10, no. 1: 162. https://doi.org/10.3390/biomedicines10010162
APA StylePalstrøm, N. B., Matthiesen, R., Rasmussen, L. M., & Beck, H. C. (2022). Recent Developments in Clinical Plasma Proteomics—Applied to Cardiovascular Research. Biomedicines, 10(1), 162. https://doi.org/10.3390/biomedicines10010162