
Characterization of the Gut Microbiota in Individuals with Overweight or Obesity during a Real-World Weight Loss Dietary Program: A Focus on the Bacteroides 2 Enterotype

 , , , and
, , , and
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Overall Dietary Program
2.1.1. Nutritional and Psycho-Behavioral Reeducation (RNPC) Program
2.1.2. The GutInside Study
2.2. Sample Collection and Bacterial DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Bioinformatics Treatment
2.5. Statistical Analyzes
3. Results
3.1. Clinical and Biological Variables of the Studied Population
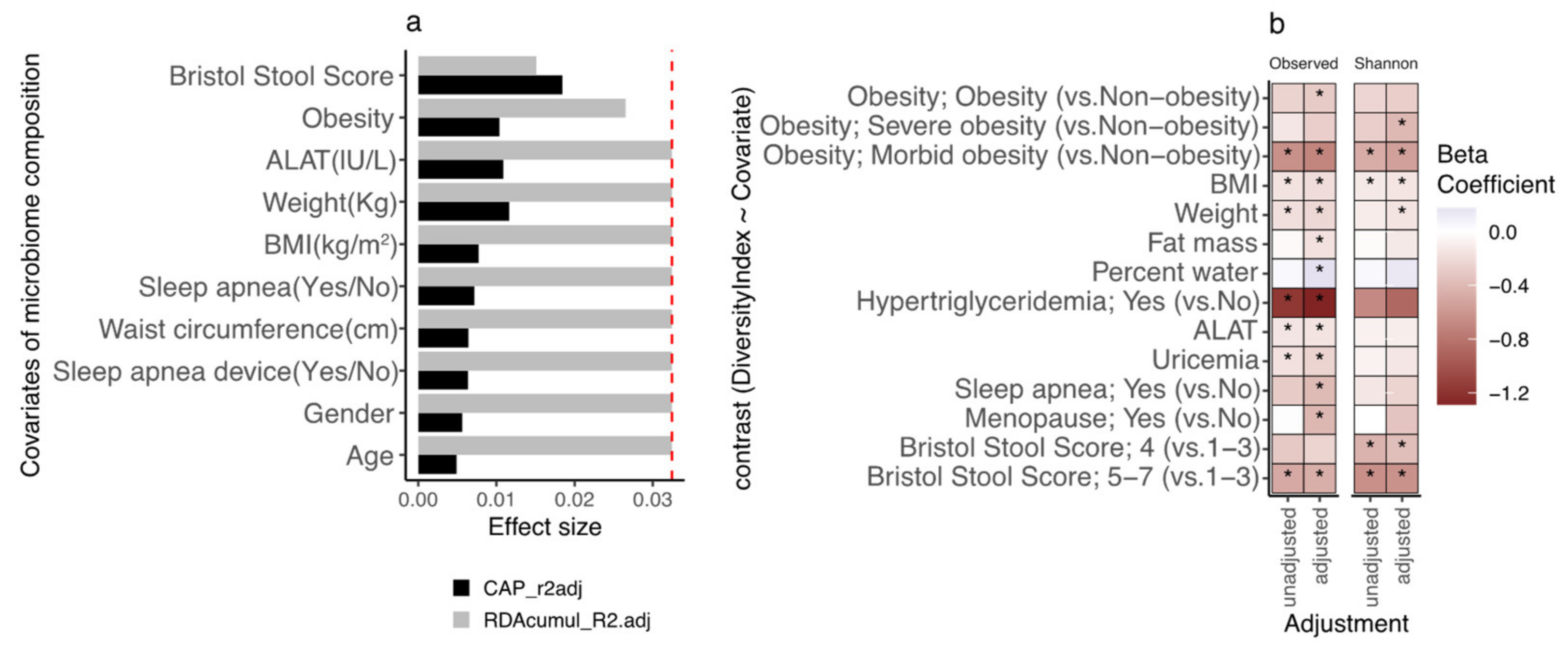
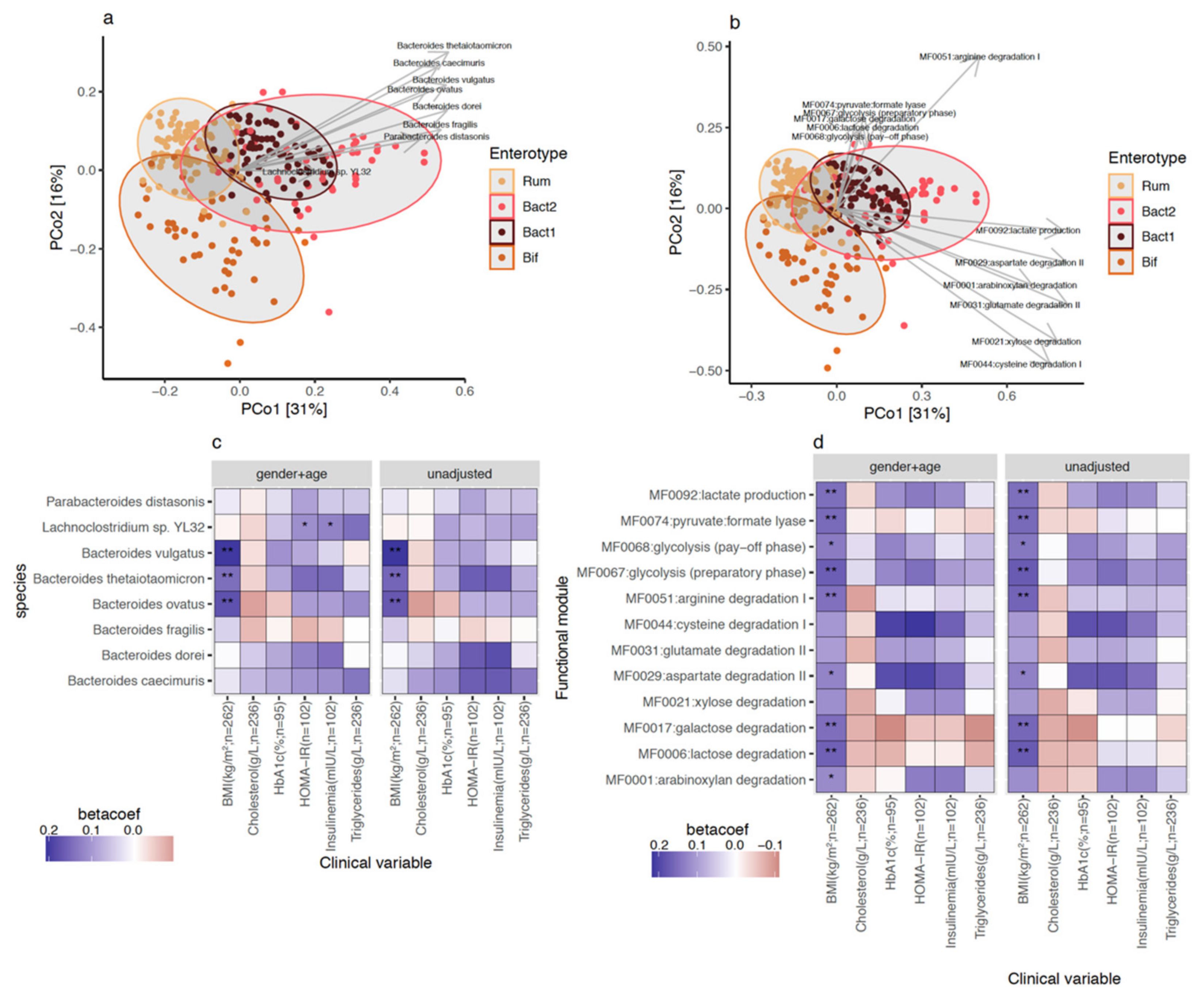
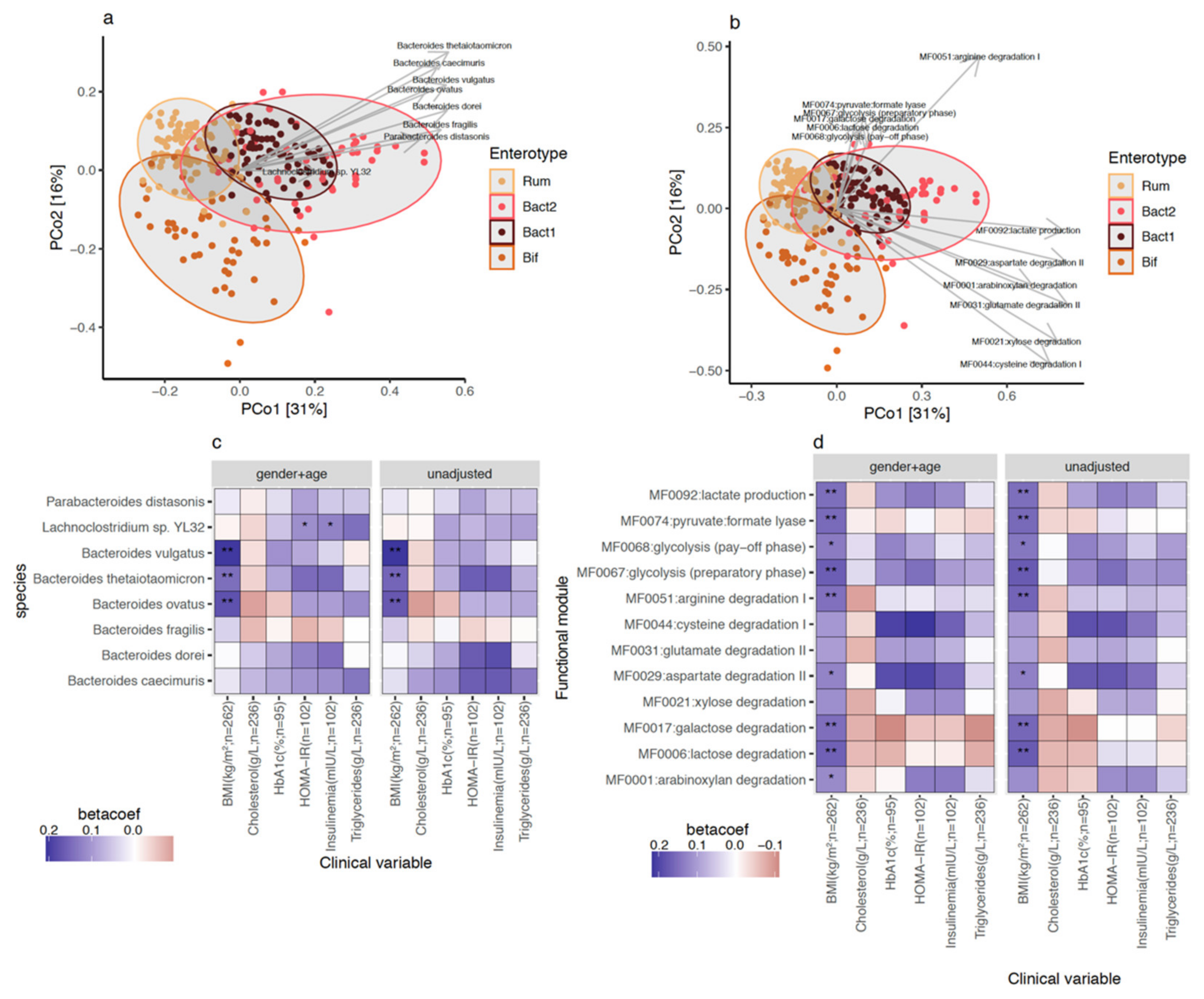
3.2. Participants’ Gut Microbial Composition, the Bact2 Enterotype, and Metabolic Profile at Baseline
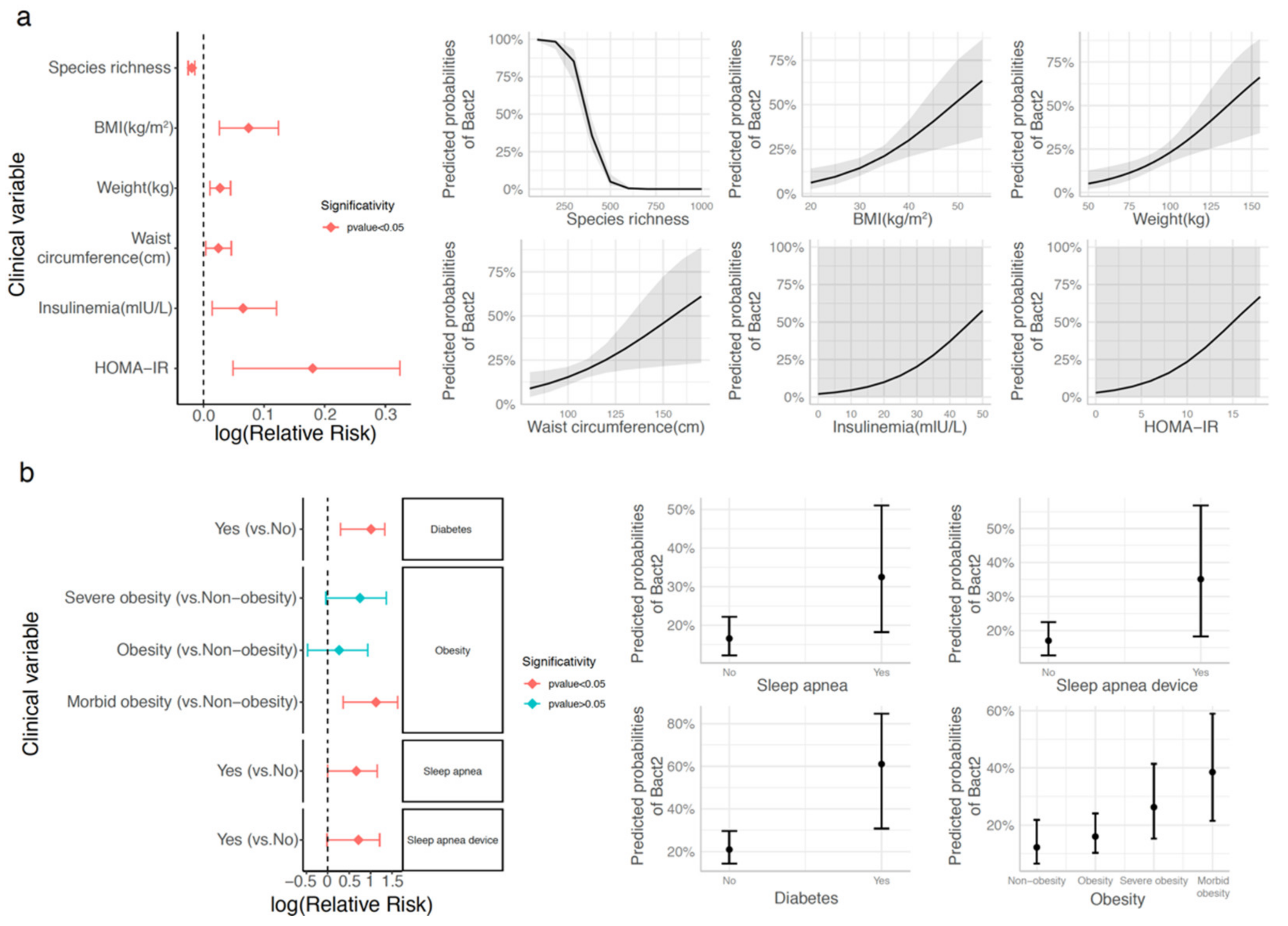
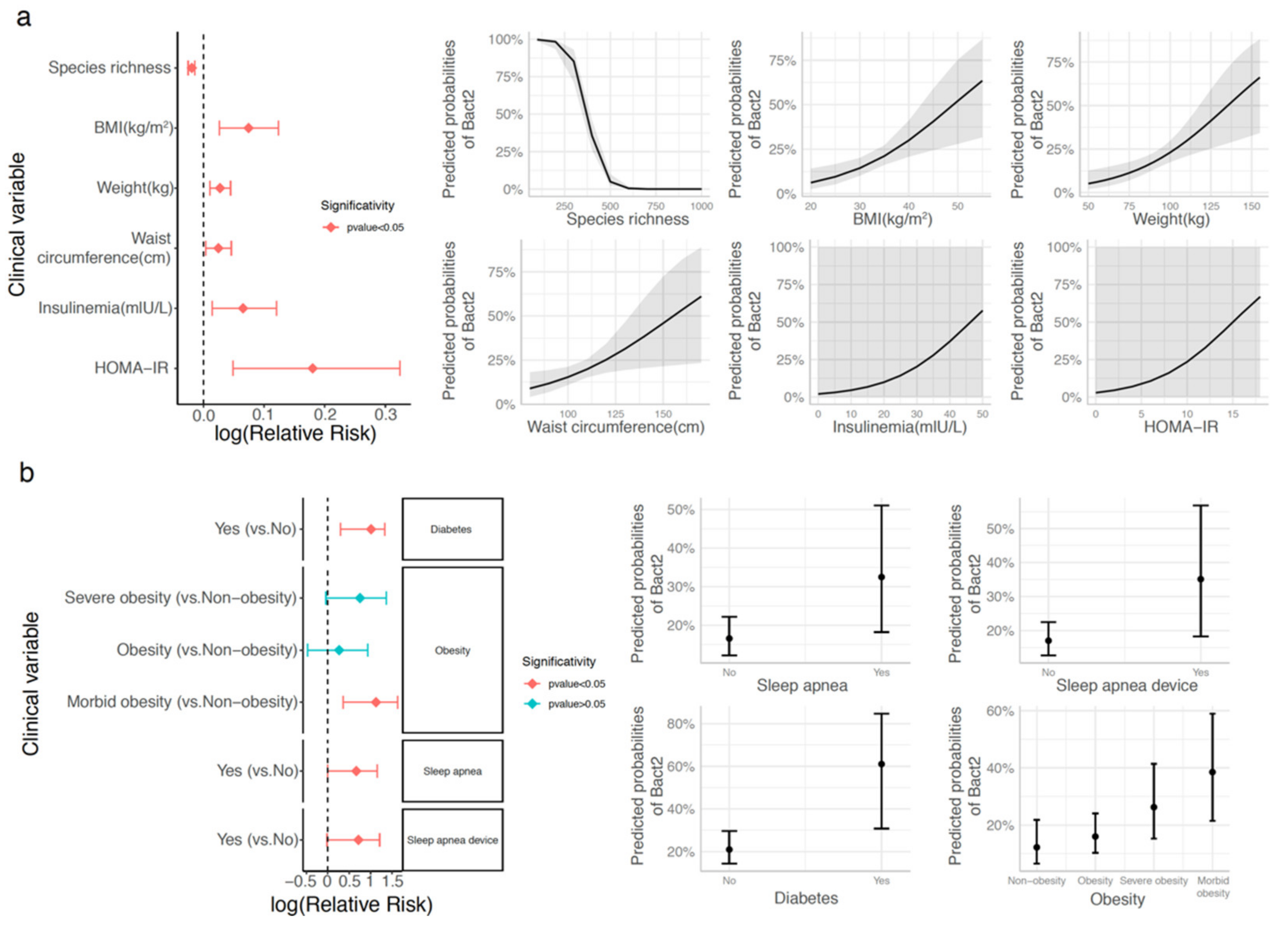
3.3. Bacteroides 2 Enterotype, Taxonomy, and Functional Features and Obesity Severity
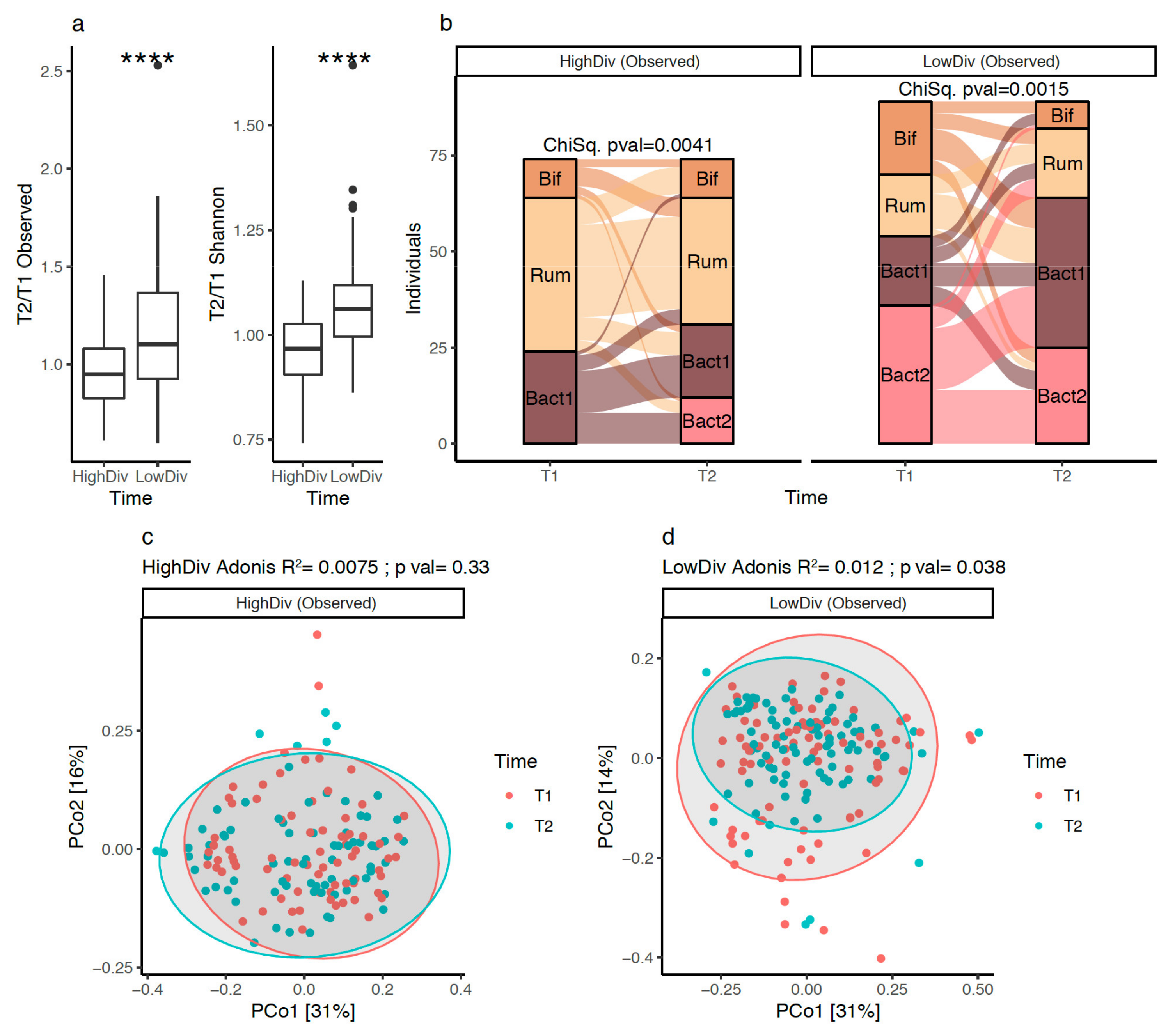
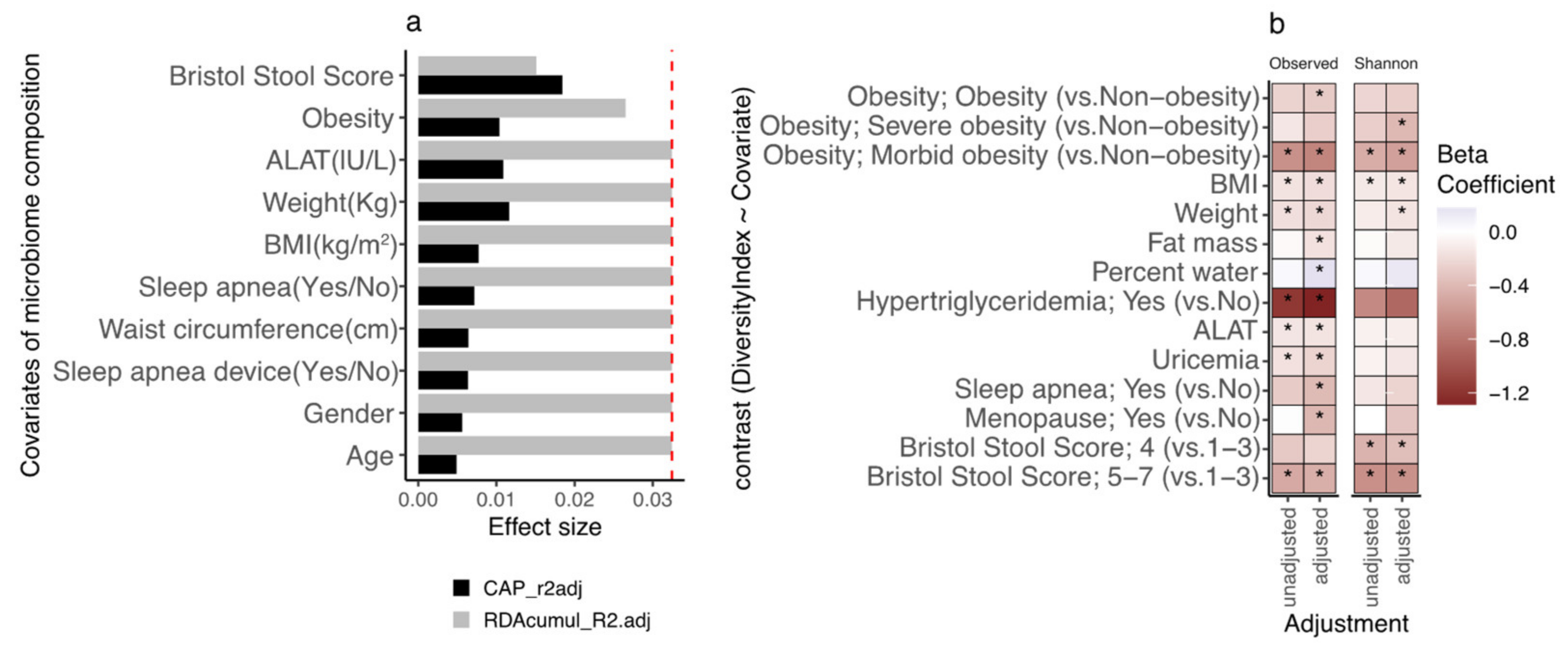
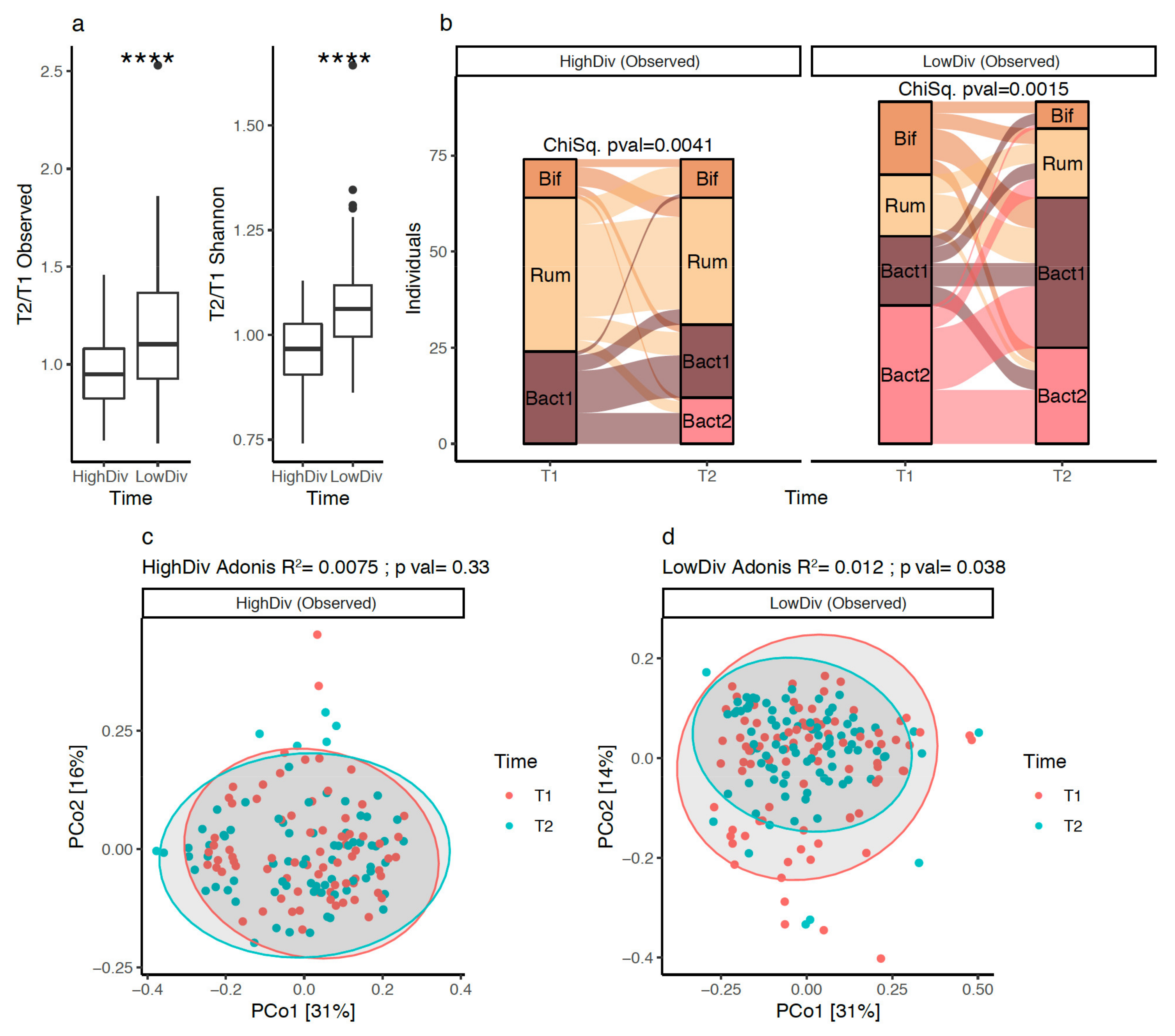
3.4. Gut Microbiota Diversity Evolution during Weight Loss Is Influenced by Baseline Gut Microbiome Diversity
3.5. Bacteroides 2 Prevalence after Weight-Loss
3.6. Bacterial Species after Weight Loss; Different Pattern with Diversity Stratification
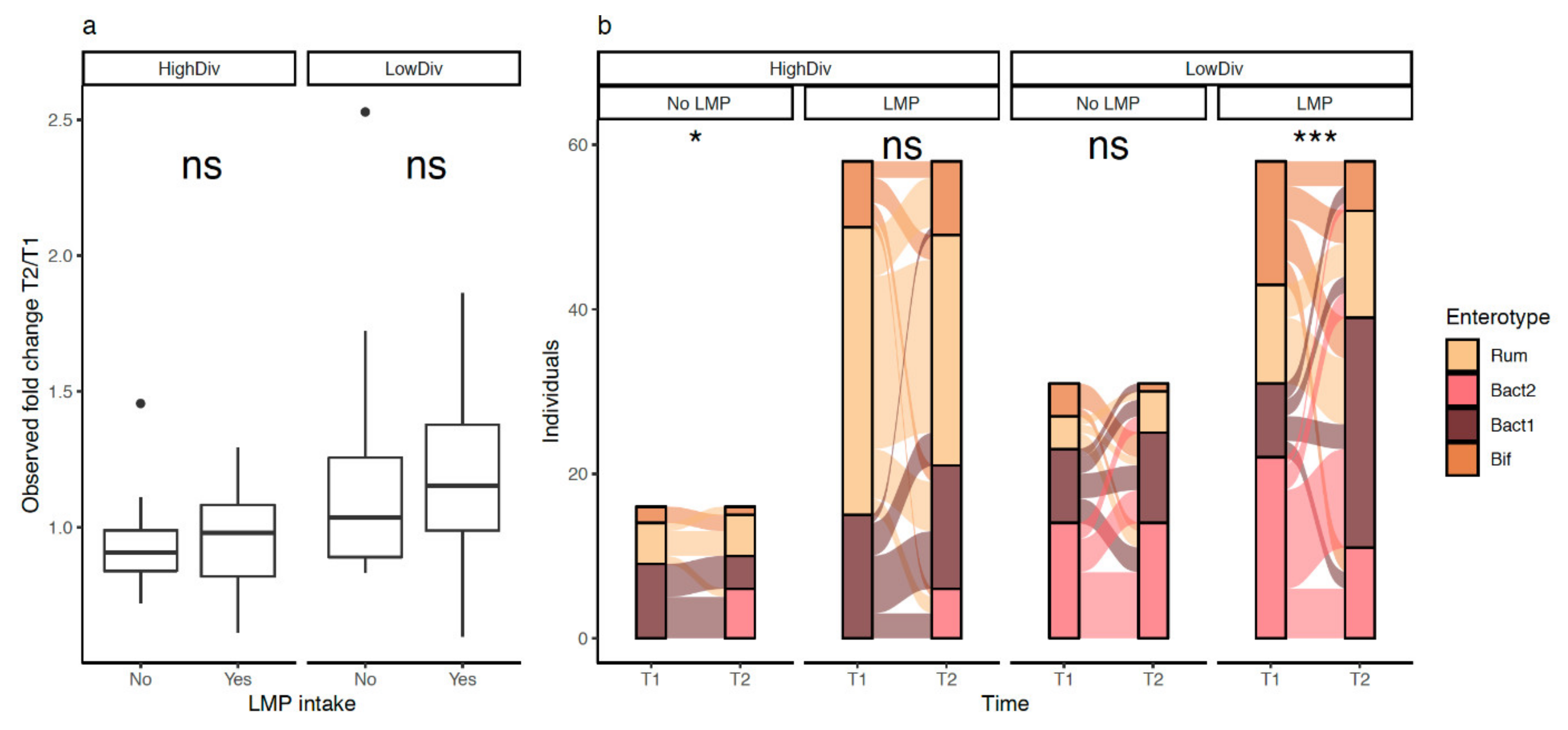
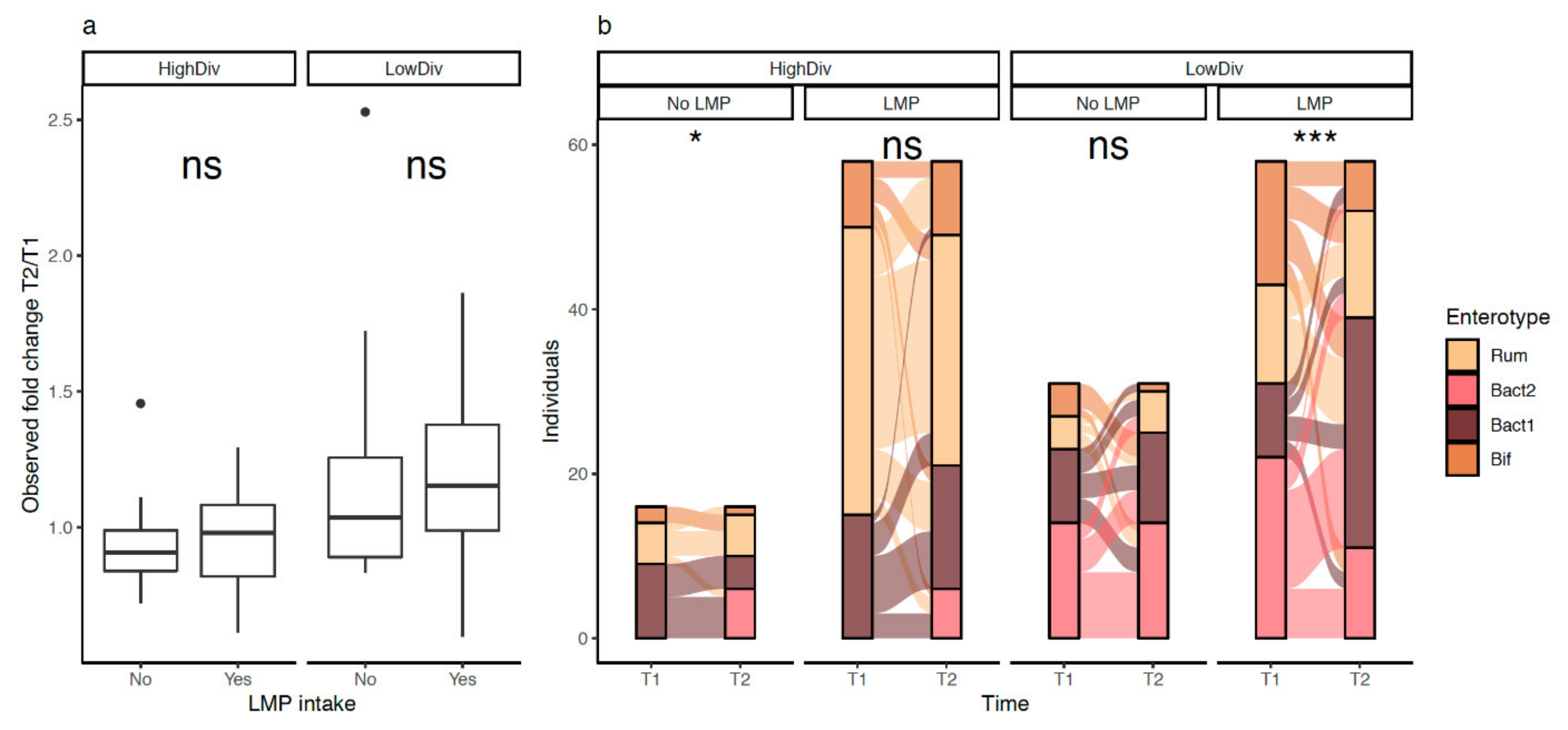
3.7. LMP Supplementation, Microbial Diversity and Composition, and Bacteroides 2 Prevalence
3.8. LMP Supplementation, Gut Microbial Species and Functional Module in Matched Subjects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 10 October 2021).
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rodriguez, E.; Guillen-Grima, F.; Martí, A.; Brugos-Larumbe, A. Comorbidity associated with obesity in a large population: The APNA study. Obes. Res. Clin. Pr. 2015, 9, 435–447. [Google Scholar] [CrossRef]
- Ryan, D.H.; Kahan, S. Guideline Recommendations for Obesity Management. Med. Clin. N. Am. 2018, 102, 49–63. [Google Scholar] [CrossRef]
- Beam, A.; Clinger, E.; Hao, L. Effect of Diet and Dietary Components on the Composition of the Gut Microbiota. Nutrients 2021, 13, 2795. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell 2015, 163, 1079–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, S.E.; Valdes, A.M.; Drew, D.A.; Asnicar, F.; Mazidi, M.; Wolf, J.; Capdevila, J.; Hadjigeorgiou, G.; Davies, R.; Al Khatib, H.; et al. Human postprandial responses to food and potential for precision nutrition. Nat. Med. 2020, 26, 964–973. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.; Hedjazil, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; De Commer, L.; Tito, R.Y.; Kathagen, G.; Sabino, J.; Vermeire, S.; Faust, K.; Raes, J. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Vandeputte, D.; Kathagen, G.; D’Hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nat. Cell Biol. 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin Therapy Is Associated with Lower Prevalence of Gut Microbiota Dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; De Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; Van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of diet to the composition of the human gut microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef]
- Dong, T.S.; Luu, K.; Lagishetty, V.; Sedighian, F.; Woo, S.-L.; Dreskin, B.W.; Katzka, W.; Chang, C.; Zhou, Y.; Arias-Jayo, N.; et al. A High Protein Calorie Restriction Diet Alters the Gut Microbiome in Obesity. Nutrients 2020, 12, 3221. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P.B.; Belda, E.; Prifti, E.; Dao, M.C.; Specque, F.; Henegar, C.; Rinaldi, L.; Wang, X.; Kennedy, S.P.; Zucker, J.-D.; et al. Protein supplementation during an energy-restricted diet induces visceral fat loss and gut microbiota amino acid metabolism activation: A randomized trial. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Kobyliak, N.; Conte, C.; Cammarota, G.; Haley, A.P.; Styriak, I.; Gaspar, L.; Fusek, J.; Rodrigo, L.; Kruzliak, P. Probiotics in prevention and treatment of obesity: A critical view. Nutr. Metab. 2016, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mistretta, A.; Volti, G.L.; et al. Bifidobacterium longum with Fructo-Oligosaccharides in Patients with Non Alcoholic Steatohepatitis. Dig. Dis. Sci. 2012, 57, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.M.; Dalskov, S.-M.; Van Baak, M.; Jebb, S.A.; Papadaki, A.; Pfeiffer, A.F.; Martinez, J.A.; Handjieva-Darlenska, T.; Kunešová, M.; Pihlsgård, M.; et al. Diets with High or Low Protein Content and Glycemic Index for Weight-Loss Maintenance. N. Engl. J. Med. 2010, 363, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Thorning, T.K.; Fabre, O.; Legrand, R.; Astrup, A.; Hjorth, M.F. Weight loss and weight loss maintenance efficacy of a novel weight loss program: The retrospective RNPC® cohort. Obes. Med. 2018, 10, 16–23. [Google Scholar] [CrossRef]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Statement on the conditions of use for health claims related to meal replacements for weight control. EFSA J. 2015, 13, 4287. [Google Scholar] [CrossRef] [Green Version]
- Alili, R.; Belda, E.; Le, P.; Wirth, T.; Zucker, J.-D.; Prifti, E.; Clément, K. Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol. Genes 2021, 12, 1496. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2019. Available online: https://cran.ism.ac.jp/web/packages/vegan/vegan.pdf (accessed on 10 October 2021).
- Holmes, I.; Harris, K.; Quince, C. Dirichlet Multinomial Mixtures: Generative Models for Microbial Metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonov, G.; Opedal, Ø.H.; Abrego, N.; Lehikoinen, A.; De Jonge, M.M.J.; Oksanen, J.; Ovaskainen, O. Joint species distribution modelling with the r -package H msc. Methods Ecol. Evol. 2019, 11, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Darzi, Y.; Falony, G.; Vieira-Silva, S.; Raes, J. Towards biome-specific analysis of meta-omics data. ISME J. 2016, 10, 1025–1028. [Google Scholar] [CrossRef] [Green Version]
- Diamond, A.; Sekhon, J.S. Genetic Matching for Estimating Causal Effects: A General Multivariate Matching Method for Achieving Balance in Observational Studies. Rev. Econ. Stat. 2013, 95, 932–945. [Google Scholar] [CrossRef] [Green Version]
- Sekhon, J.S. Multivariate and Propensity Score Matching Software with Automated Balance Optimization: The Matching Package for R. J. Stat. Softw. 2011, 42, 1–52. [Google Scholar] [CrossRef] [Green Version]
- Knights, D.; Ward, T.L.; McKinlay, C.E.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Rethinking “Enterotypes”. Cell Host Microbe 2014, 16, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Ning, K. Stereotypes About Enterotype: The Old and New Ideas. Genom. Proteom. Bioinform. 2019, 17, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Reynders, T.; Devolder, L.; Valles-Colomer, M.; Van Remoortel, A.; Joossens, M.; De Keyser, J.; Nagels, G.; D’Hooghe, M.; Raes, J. Gut microbiome variation is associated to Multiple Sclerosis phenotypic subtypes. Ann. Clin. Transl. Neurol. 2020, 7, 406–419. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Darzi, Y.; Lima-Mendez, G.; Yunta, R.G.; Okuda, S.; Vandeputte, D.; Valles-Colomer, M.; Hildebrand, F.; Chaffron, S.; et al. Species–function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol. 2016, 1, 16088. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Duncan, S.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium prausnitzii: From microbiology to diagnostics and prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef]
- Romo-Vaquero, M.; Martín, A.C.; Loria-Kohen, V.; Ramírez-De-Molina, A.; García-Mantrana, I.; Collado, M.C.; Espín, J.C.; Selma, M.V. Deciphering the Human Gut Microbiome of Urolithin Metabotypes: Association with Enterotypes and Potential Cardiometabolic Health Implications. Mol. Nutr. Food Res. 2019, 63, e1800958. [Google Scholar] [CrossRef]
- Molinaro, A.; The MetaCardis Consortium; Lassen, P.B.; Henricsson, M.; Wu, H.; Adriouch, S.; Belda, E.; Chakaroun, R.; Nielsen, T.; Bergh, P.-O.; et al. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mobeen, F.; Sharma, V.; Prakash, T. Enterotype Variations of the Healthy Human Gut Microbiome in Different Geographical Regions. Bioinformation 2018, 14, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.C.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: The NU-AGE 1-year dietary intervention across five European countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, D.; Whelan, K.; Rossi, M.; Morrison, M.; Holtmann, G.; Kelly, J.T.; Shanahan, E.R.; Staudacher, H.M.; Campbell, K.L. Dietary fiber intervention on gut microbiota composition in healthy adults: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2018, 107, 965–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karl, J.P.; Meydani, M.; Barnett, J.B.; Vanegas, S.M.; Goldin, B.; Kane, A.; Rasmussen, H.; Saltzman, E.; Vangay, P.; Knights, D.; et al. Substituting whole grains for refined grains in a 6-wk randomized trial favorably affects energy-balance metrics in healthy men and postmenopausal women. Am. J. Clin. Nutr. 2017, 105, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Sanz, Y. Effects of a gluten-free diet on gut microbiota and immune function in healthy adult humans. Gut Microbes 2010, 1, 135–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2010, 5, 220–230. [Google Scholar] [CrossRef]
- Salonen, A.H.; Lahti, L.; Salojärvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.; Date, P.; Farquharson, F.; Johnstone, A.; Lobley, G.E.; et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.; Portune, K.J.; Steuer, N.; Lan, A.; Cerrudo, V.; Audebert, M.; Dumont, F.; Mancano, G.; Khodorova, N.; Andriamihaja, M.; et al. Quantity and source of dietary protein influence metabolite production by gut microbiota and rectal mucosa gene expression: A randomized, parallel, double-blind trial in overweight humans. Am. J. Clin. Nutr. 2017, 106, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.; Flint, H.J.; Lobley, G.E. Reduced Dietary Intake of Carbohydrates by Obese Subjects Results in Decreased Concentrations of Butyrate and Butyrate-Producing Bacteria in Feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Cani, P.D.; Neyrinck, A.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.S.; Gibson, G.R.; Hutkins, R.; Reimer, R.A.; Reid, G.; Verbeke, K.; Scott, K.P.; Holscher, H.D.; Azad, M.B.; Delzenne, N.M.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of synbiotics. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Dewulf, E.M.; Cani, P.; Claus, S.; Fuentes, S.; Puylaert, P.G.B.; Neyrinck, A.; Bindels, L.B.; De Vos, W.M.; Gibson, G.R.; Thissen, J.-P.; et al. Insight into the prebiotic concept: Lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 2012, 62, 1112–1121. [Google Scholar] [CrossRef]
- Wang, K.; Liao, M.; Zhou, N.; Bao, L.; Ma, K.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, W.; Wang, J.; et al. Parabacteroides distasonis Alleviates Obesity and Metabolic Dysfunctions via Production of Succinate and Secondary Bile Acids. Cell Rep. 2019, 26, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Bel Lassen, P.B.; Attaye, I.; Adriouch, S.; Nicolaou, M.; Aron-Wisnewsky, J.; Nielsen, T.; Chakaroun, R.; Le Chatelier, E.; Forslund, S.; Belda, E.; et al. Protein Intake, Metabolic Status and the Gut Microbiota in Different Ethnicities: Results from Two Independent Cohorts. Nutrients 2021, 13, 3159. [Google Scholar] [CrossRef] [PubMed]
- Al Bataineh, M.T.; Dash, N.; Lassen, P.B.; Banimfreg, B.H.; Nada, A.M.; Belda, E.; Clément, K. Revealing links between gut microbiome and its fungal community in Type 2 Diabetes Mellitus among Emirati subjects: A pilot study. Sci. Rep. 2020, 10, 9624. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.P.; Moreto, F.; Silveira, L.; Burini, R.C. Dietary, anthropometric, and biochemical determinants of uric acid in free-living adults. Nutr. J. 2013, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, F.; Zhang, C.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; et al. Gut bacteria selectively promoted bydietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeev, I.N.; Aljutaily, T.; Walton, G.; Huarte, E. Effects of Synbiotic Supplement on Human Gut Microbiota, Body Composition and Weight Loss in Obesity. Nutrition 2020, 12, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | All | No LMP | LMP | p-Value |
|---|---|---|---|---|
| Sample size, n (%) | 263 (100.0) | 75 (28.5) | 188 (71.5) | - |
| Age (years) | 50.6 (9.9) | 50.8 (10.1) | 50.5 (9.8) | ns |
| Sex (male), n (%) | 55 (20.9) | 21 (28.0) | 34 (18.1) | ns |
| Height (cm) | 165.8 (7.9) | 167.2 (8.4) | 165.3 (7.7) | ns |
| Adiposity markers | ||||
| Weight (kg) | 90.5 (16.3) | 93.1 (18.4) | 89.4 (15.3) | ns |
| BMI (kg/m2) | 32.8 (4.9) | 33.2 (5.4) | 32.7 (4.8) | ns |
| Waist circumference (cm) | 106.5 (12.4) | 107.9 (13.4) | 106.0 (12.0) | ns |
| Fat mass (%) | 40.7 (4.7) | 40.3 (4.7) | 40.9 (4.8) | ns |
| Fat-free mass (%) | 32.0 (7.5) | 30.8 (5.6) | 32.0 (8.1) | ns |
| Calculated Basal metabolism (Kcal/day) | 2217 (317) | 2281 (363) | 2192 (294) | ns |
| Plasma Glucose homeostasis | ||||
| Glycemia (g/L) | 1.02 (0.22) | 1.06 (0.23) | 1.01 (0.21) | * |
| Insulinemia (mIU/L) | 14.9 (8.7) | 16.6 (10.2) | 14.3 (8.0) | ns |
| HOMA IR | 4.1 (3.1) | 4.9 (4.1) | 3.5 (2.7) | ns |
| HbA1c (%) | 5.8 (0.9) | 6.0 (0.9) | 5.8 (0.9) | ns |
| Plasma lipid homeostasis | ||||
| Total cholesterol (nmol/L) | 5.5 (1.1) | 5.6 (1.0) | 5.5 (1.1) | ns |
| Total triglycerides (nmol/L) | 1.6 (0.9) | 1.7 (1.2) | 1.5 (0.8) | ns |
| HDL cholesterol (nmol/L) | 1.4 (0.4) | 1.4 (0.4) | 1.4 (0.4) | ns |
| LDL cholesterol (nmol/L) | 3.4 (1.0) | 3.5 (0.8) | 3.4 (1.1) | ns |
| Liver Enzymes | ||||
| ASAT (IU/L) | 24.0 (10.0) | 24.0 (9.7) | 24.0 (10.2) | ns |
| ALAT (IU/L) | 30.2 (18.3) | 33.1 (20.7) | 29.1 (17.1) | ns |
| GGT (IU/L) | 41.0 (40.9) | 54.3 (50.1) | 36.0 (36.0) | * |
| Other variables | ||||
| Creatinine (mg/L) | 8.1 (1.5) | 8.1 (1.6) | 8.1 (1.5) | ns |
| Uricemia (mg/L) | 52.1 (16.3) | 55.1 (11.3) | 51.0 (17.6) | ns |
| SAS (Yes), n (%) | 37 (14.1) | 10 (13.3) | 27 (14.4) | ns |
| Clinical Parameters | Before Weight Loss | After Weight Loss (-10%) | p-Value |
|---|---|---|---|
| Sample size, n (%) | 163 (100.0) | - | - |
| Age (years) | 50.8 (10.1) | - | - |
| Sex (male), n (%) | 31 (19.0) | - | - |
| Height (cm) | 165.4 (7.8) | - | - |
| SAS (Yes), n (%) | 25 (15.3) | - | - |
| Adiposity markers | |||
| Weight (kg) | 90.4 (16.1) | 80.4 (14.4) | **** |
| BMI (kg/m2) | 33.0 (4.9) | 29.3 (4.4) | **** |
| Waist circumference (cm) | 106.9 (12.5) | 96.3 (11.7) | **** |
| Fat mass (%) | 40.8 (4.4) | 37.7 (5.0) | **** |
| Fat-free mass (%) | 32.5 (8.3) | 33.2 (7.6) | **** |
| Calculated Basal metabolism (Kcal/day) | 2195 (306) | 2080 (295) | **** |
| Plasma Glucose homeostasis | |||
| Glycemia (g/L) | 1.03 (0.20) | 0.97 (0.12) | *** |
| Insulinemia (mIU/L) | 14.0 (7.6) | 10.2 (6.3) | ** |
| HOMA IR | 4.3 (3.7) | 2.5 (1.9) | * |
| HbA1c (%) | 6.2 (1.1) | 5.6 (0.7) | ** |
| Plasma lipid homeostasis | |||
| Total cholesterol (nmol/L) | 5.7 (1.0) | 5.1 (1.0) | **** |
| Total triglycerides (nmol/L) | 1.6 (0.8) | 1.1 (0.5) | **** |
| HDL cholesterol (nmol/L) | 1.4 (0.4) | 1.4 (0.4) | ns |
| LDL cholesterol (nmol/L) | 3.5 (0.9) | 3.2 (0.8) | **** |
| Liver Enzymes | |||
| ASAT (IU/L) | 23.8 (8.5) | 21.9 (7.6) | ** |
| ALAT (IU/L) | 30.0 (18.0) | 24.6 (12.1) | ** |
| GGT (IU/L) | 50.3 (53.4) | 34.3 (42.2) | ** |
| Other variables | |||
| Creatinine (mg/L) | 8.4 (1.5) | 8.3 (1.4) | ns |
| Uricemia (mg/L) | 53.0 (16.0) | 49.8 (15.1) | ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alili, R.; Belda, E.; Fabre, O.; Pelloux, V.; Giordano, N.; Legrand, R.; Bel Lassen, P.; Swartz, T.D.; Zucker, J.-D.; Clément, K. Characterization of the Gut Microbiota in Individuals with Overweight or Obesity during a Real-World Weight Loss Dietary Program: A Focus on the Bacteroides 2 Enterotype. Biomedicines 2022, 10, 16. https://doi.org/10.3390/biomedicines10010016
Alili R, Belda E, Fabre O, Pelloux V, Giordano N, Legrand R, Bel Lassen P, Swartz TD, Zucker J-D, Clément K. Characterization of the Gut Microbiota in Individuals with Overweight or Obesity during a Real-World Weight Loss Dietary Program: A Focus on the Bacteroides 2 Enterotype. Biomedicines. 2022; 10(1):16. https://doi.org/10.3390/biomedicines10010016
Chicago/Turabian StyleAlili, Rohia, Eugeni Belda, Odile Fabre, Véronique Pelloux, Nils Giordano, Rémy Legrand, Pierre Bel Lassen, Timothy D. Swartz, Jean-Daniel Zucker, and Karine Clément. 2022. "Characterization of the Gut Microbiota in Individuals with Overweight or Obesity during a Real-World Weight Loss Dietary Program: A Focus on the Bacteroides 2 Enterotype" Biomedicines 10, no. 1: 16. https://doi.org/10.3390/biomedicines10010016
APA StyleAlili, R., Belda, E., Fabre, O., Pelloux, V., Giordano, N., Legrand, R., Bel Lassen, P., Swartz, T. D., Zucker, J.-D., & Clément, K. (2022). Characterization of the Gut Microbiota in Individuals with Overweight or Obesity during a Real-World Weight Loss Dietary Program: A Focus on the Bacteroides 2 Enterotype. Biomedicines, 10(1), 16. https://doi.org/10.3390/biomedicines10010016