
Endurance Training in Humans Modulates the Bacterial DNA Signature of Skeletal Muscle


Abstract
:1. Background
2. Methods
2.1. Participants, Training Protocol and Sample Collection
2.2. Bacterial DNA Extraction
2.3. Negative Controls
2.4. 16S rRNA Gene Amplicon Sequencing
2.5. Clustering
2.6. Comparing Taxa Abundance before and after Training
2.7. Correlation between OTU Counts and Clinical Parameters
- Alpha distributions vs. clinical parameters
- Δ (posttraining-pretraining) alpha distribution vs. Δ (posttraining-pretraining) clinical parameters
- Filtered OTUs counts vs. clinical parameters
- Δ posttraining-pretraining) filtered OTUs counts vs. Δ (posttraining-pretraining) clinical parameters.
2.8. Visualisation
3. Results
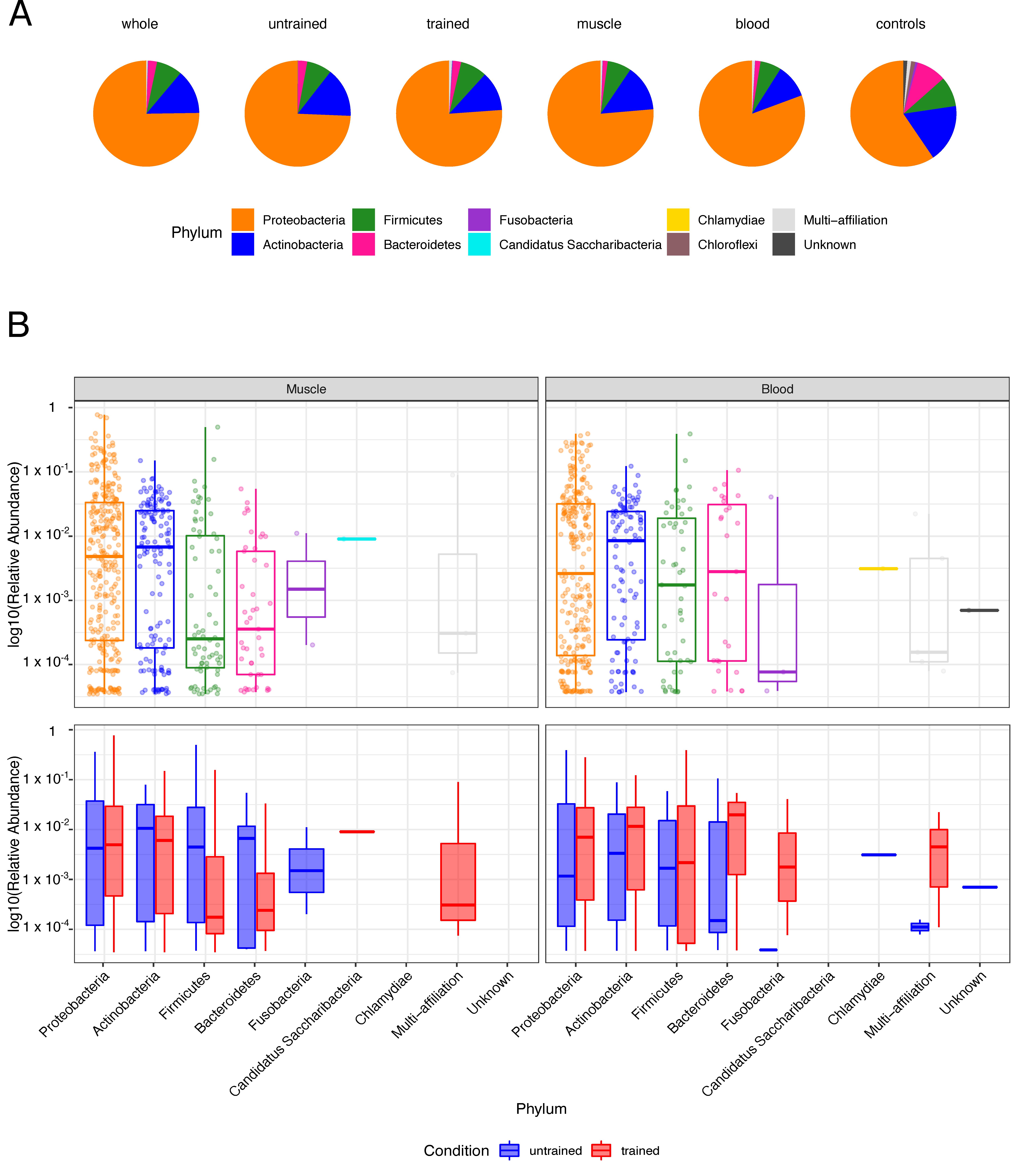
3.1. Bacterial DNA Is Present in Human Muscle and Blood
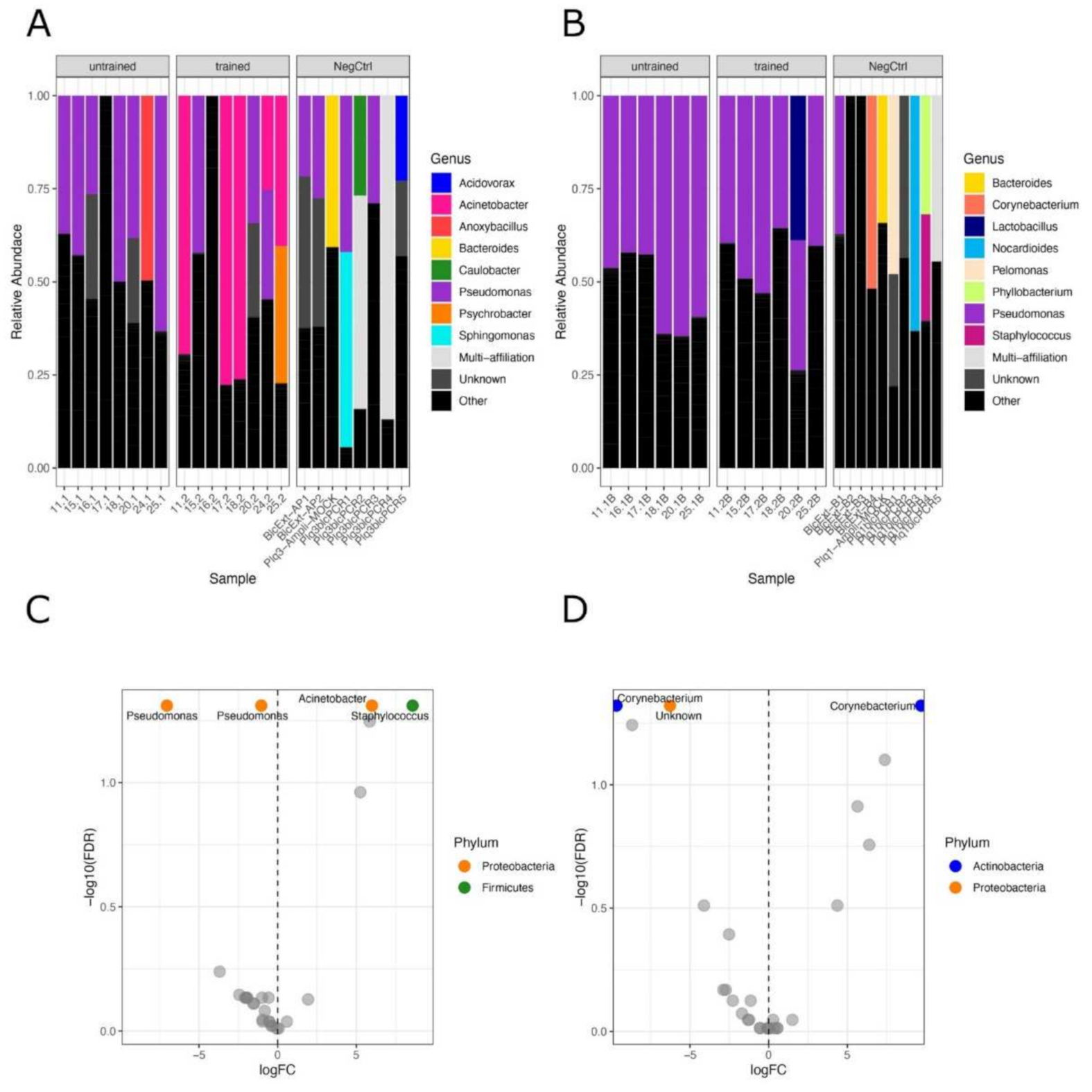
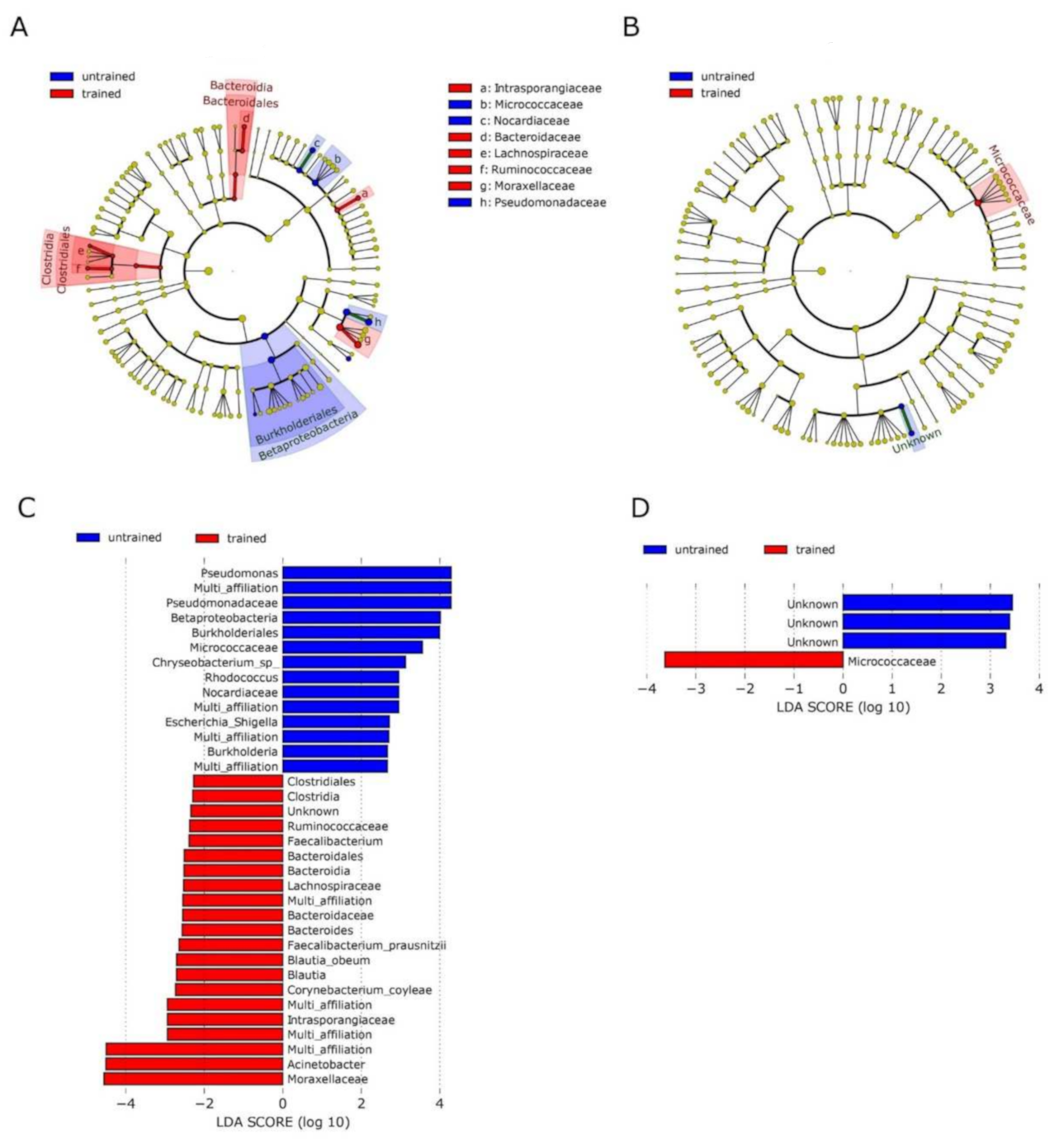
3.2. Exercise Remodels Bacterial DNA Content in the Skeletal Muscle, but Not in the Blood
3.3. No Correlation between OTUs Profiles and Clinical Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| T2D | Type 2 Diabetes |
| IL | InterLeukin |
| Th17 | T-helper cell 17 |
| rRNA | ribosomal RNA |
| qPCR | quantitative Polymerase Chain Reaction |
| OTU | Operational Taxonomic Unit |
| LEfSe | Linear discriminant analysis Effect Size |
| PCoA | Principal Coordinate Analysis |
| IP | Intestinal Permeability |
| ROS | Reactive Oxygen Species |
| GI | GastroIntestinal |
| TNF | Tumor Necrosis Factor |
| IFN | InterFeroN |
| SCFA | Short Chain Fatty-Acid |
| TCR | T-Cell Receptor |
References
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermudez-Humaran, L.G.; Smirnova, N.; Berge, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; von Bergen, M.; Haange, S.B.; Heyne, H.; Stumvoll, M.; et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 2020, 69, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Serino, M.; Chabo, C.; Garidou, L.; Pomié, C.; Courtney, M.; Amar, J.; Bouloumié, A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes Obes. Metab. 2013, 15, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, R.W.; Vali, H.; Lau, P.C.; Palfree, R.G.; De Ciccio, A.; Sirois, M.; Ahmad, D.; Villemur, R.; Desrosiers, M.; Chan, E.C. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J. Clin. Microbiol. 2002, 40, 4771–4775. [Google Scholar] [CrossRef] [Green Version]
- Costantini, L.; Magno, S.; Albanese, D.; Donati, C.; Molinari, R.; Filippone, A.; Masetti, R.; Merendino, N. Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci. Rep. 2018, 8, 16893. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.M.; Gloor, G.B.; Baban, C.K.; Scott, L.; O’Hanlon, D.M.; Burton, J.P.; Francis, K.P.; et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 2014, 80, 3007–3014. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Morris, A.; Huang, L.; Beck, J.M.; Twigg, H.L., 3rd; von Mutius, E.; Ghedin, E. The microbiome and the lung. Ann. Am. Thorac. Soc. 2014, 11, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef]
- Huang, Y.J.; Charlson, E.S.; Collman, R.G.; Colombini-Hatch, S.; Martinez, F.D.; Senior, R.M. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am. J. Respir. Crit. Care Med. 2013, 187, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal. Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.E.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The Liver May Act as a Firewall Mediating Mutualism Between the Host and Its Gut Commensal Microbiota. Sci. Transl. Med. 2014, 6, 237ra66. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Burcelin, R.; Tremaroli, V. Liver tissue microbiome in NAFLD: Next step in understanding the gut–liver axis? Gut 2020, 69, 1373–1374. [Google Scholar] [CrossRef]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-liver axis at the frontier of host-microbial interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Jensen, B.A.H.; Varin, T.V.; Servant, F.; Van Blerk, S.; Richard, D.; Marceau, S.; Surette, M.; Biertho, L.; Lelouvier, B.; et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020, 2, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Zhou, L.; Zhang, J.; Wang, B. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int. J. Clin. Exp. Pathol. 2015, 8, 5153–5160. [Google Scholar]
- Awad, W.A.; Hess, C.; Hess, M. Enteric Pathogens and Their Toxin-Induced Disruption of the Intestinal Barrier through Alteration of Tight Junctions in Chickens. Toxins 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Abdelhamid, L.; Luo, X.M. Retinoic Acid, Leaky Gut, and Autoimmune Diseases. Nutrients 2018, 10, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Q.; Kirby, J.; Reilly, C.M.; Luo, X.M. Leaky Gut as a Danger Signal for Autoimmune Diseases. Front. Immunol. 2017, 8, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Garidou, L.; Pomie, C.; Klopp, P.; Waget, A.; Charpentier, J.; Aloulou, M.; Giry, A.; Serino, M.; Stenman, L.; Lahtinen, S.; et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORgammat and Controls Metabolic Disease. Cell Metab. 2015, 22, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Lluch, J.; Servant, F.; Paisse, S.; Valle, C.; Valiere, S.; Kuchly, C.; Vilchez, G.; Donnadieu, C.; Courtney, M.; Burcelin, R.; et al. The Characterization of Novel Tissue Microbiota Using an Optimized 16S Metagenomic Sequencing Pipeline. PLoS ONE 2015, 10, e0142334. [Google Scholar]
- Lelouvier, B.; Servant, F.; Païssé, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef]
- Nay, K.; Jollet, M.; Goustard, B.; Baati, N.; Vernus, B.; Pontones, M.; Lefeuvre-Orfila, L.; Bendavid, C.; Rue, O.; Mariadassou, M.; et al. Gut bacteria are critical for optimal muscle function: A potential link with glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E158–E171. [Google Scholar] [CrossRef]
- Motiani, K.K.; Collado, M.C.; Eskelinen, J.J.; Virtanen, K.A.; Loyttyniemi, E.; Salminen, S.; Nuutila, P.; Kalliokoski, K.K.; Hannukainen, J.C. Exercise Training Modulates Gut Microbiota Profile and Improves Endotoxemia. Med. Sci. Sports Exerc. 2020, 52, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohail, M.U.; Yassine, H.M.; Sohail, A.; Al Thani, A.A. Impact of Physical Exercise on Gut Microbiome, Inflammation, and the Pathobiology of Metabolic Disorders. Rev. Diabet. Stud. 2019, 15, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Ingerslev, L.R.; Donkin, I.; Fabre, O.; Versteyhe, S.; Mechta, M.; Pattamaprapanont, P.; Mortensen, B.; Krarup, N.T.; Barrès, R. Endurance training remodels sperm-borne small RNA expression and methylation at neurological gene hotspots. Clin. Epigenet. 2018, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Pattamaprapanont, P.; Garde, C.; Fabre, O.; Barrès, R. Muscle Contraction Induces Acute Hydroxymethylation of the Exercise-Responsive Gene Nr4a3. Front. Endocrinol. 2016, 7, 165. [Google Scholar] [CrossRef] [Green Version]
- Fabre, O.; Ingerslev, L.R.; Garde, C.; Donkin, I.; Simar, D.; Barrès, R. Exercise training alters the genomic response to acute exer-cise in human adipose tissue. Epigenomics 2018, 10, 1033–1050. [Google Scholar] [CrossRef] [Green Version]
- Schierwagen, R.; Alvarez-Silva, M.C.; Servant, F.; Trebicka, J.; Lelouvier, B.; Arumugam, M. Trust is good, control is better: Technical considerations in blood microbiome analysis. Gut 2019, 69, 1362–1363. [Google Scholar] [CrossRef] [PubMed]
- Mahé, F.; Rognes, T.; Quince, C.; De Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential Expresssion Analysis of Multifactor RNA-Seq Experiments with Respect to Biological Variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood micro-biota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Chen, B.; Yang, J.; Wang, J.; Zhu, D.; Meng, Q.; Zhang, L. Study of Microbiomes in Aseptically Collected Samples of Human Breast Tissue Using Needle Biopsy and the Potential Role of in situ Tissue Microbiomes for Promoting Malignancy. Front. Oncol. 2018, 8, 318. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.C.; Farmer, C.B.; Walker, C.K. The human brain microbiome; there are bacteria in our brains! Soc. Neurosci. 2018, 2018, 594. [Google Scholar]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Entero-types of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.D.; Jung, M.J.; Roh, S.W.; Kim, M.S.; Bae, J.W. Comparative analysis of Korean human gut microbiota by barcoded pyrose-quencing. PLoS ONE 2011, 6, e22109. [Google Scholar] [CrossRef]
- Perez-Chaparro, P.J.; Goncalves, C.; Figueiredo, L.C.; Faveri, M.; Lobao, E.; Tamashiro, N.; Duarte, P.; Feres, M. Newly identified pathogens associated with periodontitis: A systematic review. J. Dent. Res. 2014, 93, 846–858. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Costa, R.J.; Snipe, R.; Camoes-Costa, V.; Scheer, V.; Murray, A. The Impact of Gastrointestinal Symptoms and Dermatological Injuries on Nutritional Intake and Hydration Status During Ultramarathon Events. Sports Med. Open 2016, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeukendrup, A.E.; Vet-Joop, K.; Sturk, A.; Stegen, J.H.; Senden, J.; Saris, W.H.; Wagenmakers, A.J. Relationship between gastro-intestinal complaints and endotoxaemia, cytokine release and the acute-phase reaction during and after a long-distance triathlon in highly trained men. Clin. Sci. 2000, 98, 47–55. [Google Scholar] [CrossRef]
- Marchbank, T.; Davison, G.; Oakes, J.R.; Ghatei, M.A.; Patterson, M.; Moyer, M.P.; Playford, R.J. The nutriceutical bovine colostrum truncates the increase in gut permeability caused by heavy exercise in athletes. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G477–G484. [Google Scholar] [CrossRef] [Green Version]
- Vanuytsel, T.; van Wanrooy, S.; Vanheel, H.; Vanormelingen, C.; Verschueren, S.; Houben, E.; Salim, R.S.; Tόth, J.; Holvoet, L.; Farré, R.; et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 2014, 63, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, J.; Liu, Z.; Chuang, C.-C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, M.; Read, A. Effects of exercise on mesenteric blood flow in man. Gut 1987, 28, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.P. Stress-induced gastrointestinal barrier dysfunction and its inflammatory effects. J. Anim. Sci. 2009, 87, E101–E108. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.M.C.; Davison, G.W.; McClean, C.M.; Murphy, M.H. A Systematic Review of the Acute Effects of Exercise on Immune and Inflammatory Indices in Untrained Adults. Sports Med. Open 2015, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Mach, N. Exercise-induced stress behavior, gut-microbiota-brain axis and diet: A systematic review for athletes. J. Int. Soc. Sports Nutr. 2016, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- van Wijck, K.; Lenaerts, K.; van Loon, L.J.; Peters, W.H.; Buurman, W.A.; Dejong, C.H. Exercise-induced splanchnic hypoperfusion results in gut dysfunction in healthy men. PLoS ONE 2011, 6, e22366. [Google Scholar] [CrossRef] [Green Version]
- Pasini, E.; Corsetti, G.; Assanelli, D.; Testa, C.; Romano, C.; Dioguardi, F.S.; Aquilani, R. Effects of chronic exercise on gut microbio-ta and intestinal barrier in human with type 2 diabetes. Minerva Med. 2019, 110, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Karper, W.B. Intestinal permeability, moderate exercise, and older adult health. Holist. Nurs. Pract. 2011, 25, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Codella, R.; Luzi, L.; Terruzzi, I. Exercise has the guts: How physical activity may positively modulate gut microbiota in chronic and immune-based diseases. Dig. Liver Dis. 2018, 50, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Berg, R.D. Bacterial translocation from the gastrointestinal tract. Trends Microbiol. 1995, 3, 149–154. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intes-tinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, K.A.; Stappenbeck, T.S. Peripheral education of the immune system by the colonic microbiota. Semin. Immunol. 2013, 25, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Walton, R.G.; Kosmac, K.; Mula, J.; Fry, C.S.; Peck, B.D.; Groshong, J.S.; Finlin, B.S.; Zhu, B.; Kern, P.A.; Peterson, C.A. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Sci. Rep. 2019, 9, 969. [Google Scholar] [CrossRef] [Green Version]
- Haas, T.; Metzger, J.; Schmitz, F.; Heit, A.; Müller, T.; Latz, E.; Wagner, H. The DNA sugar backbone 2' deoxyribose determines toll-like receptor 9 activation. Immunity 2008, 28, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Kapoor, A.; Kaneko, M.; Smolenski, R.T.; D’Acquisto, F.; Coppen, S.R.; Harada-Shoji, N.; Lee, H.J.; Thiemermann, C.; Ta-kashima, S.; et al. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc. Natl. Acad. Sci. USA 2013, 26, 14–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Shintani, Y.; Drexler, H.C.; Kioka, H.; Terracciano, C.M.; Coppen, S.R.; Imamura, H.; Akao, M.; Nakai, J.; Wheeler, A.P.; Higo, S.; et al. Toll-like receptor 9 protects non-immune cells from stress by modulating mi-tochondrial ATP synthesis through the inhibition of SERCA2. EMBO Rep. 2014, 15, 45–438. [Google Scholar] [CrossRef] [PubMed]
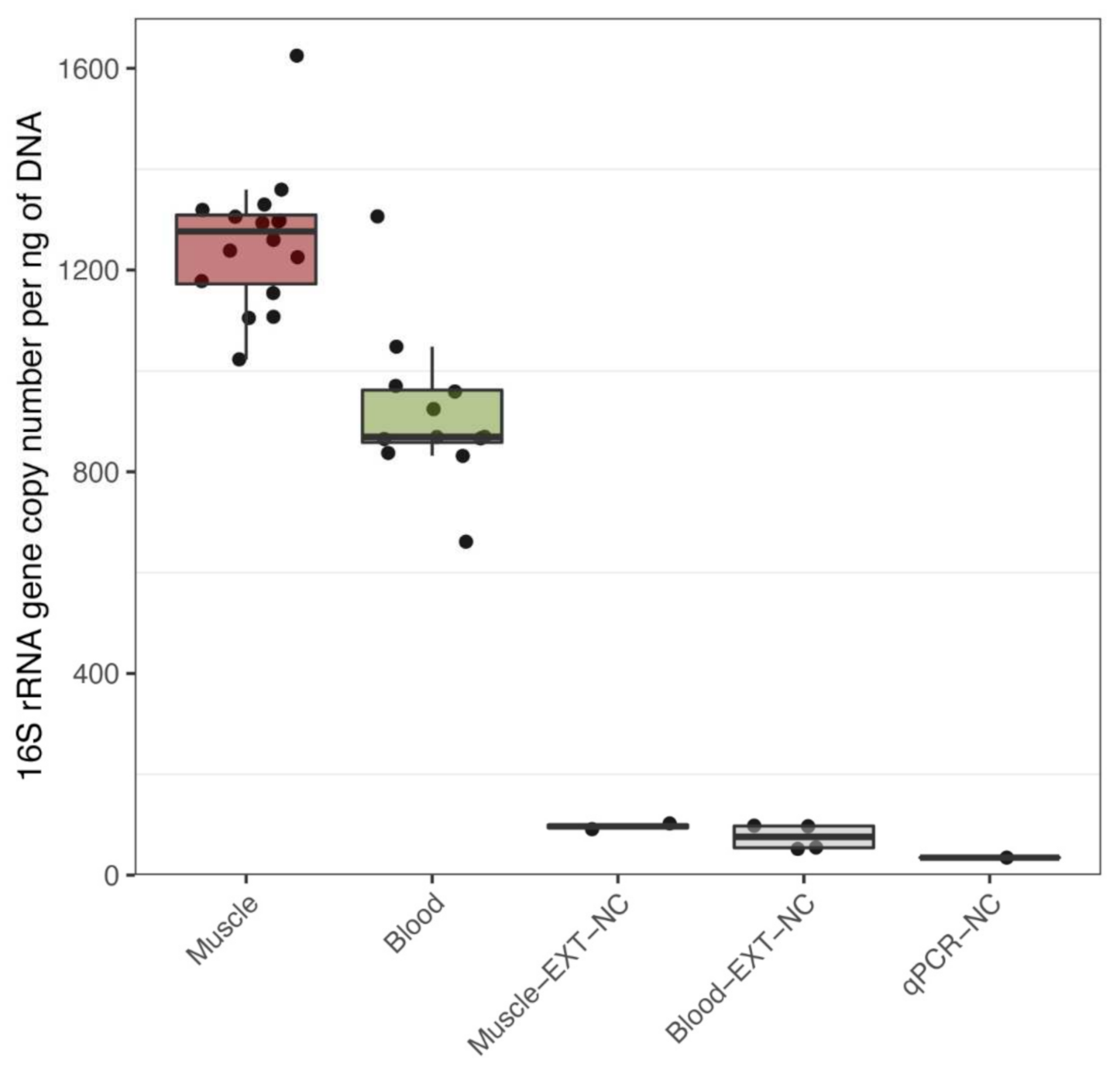
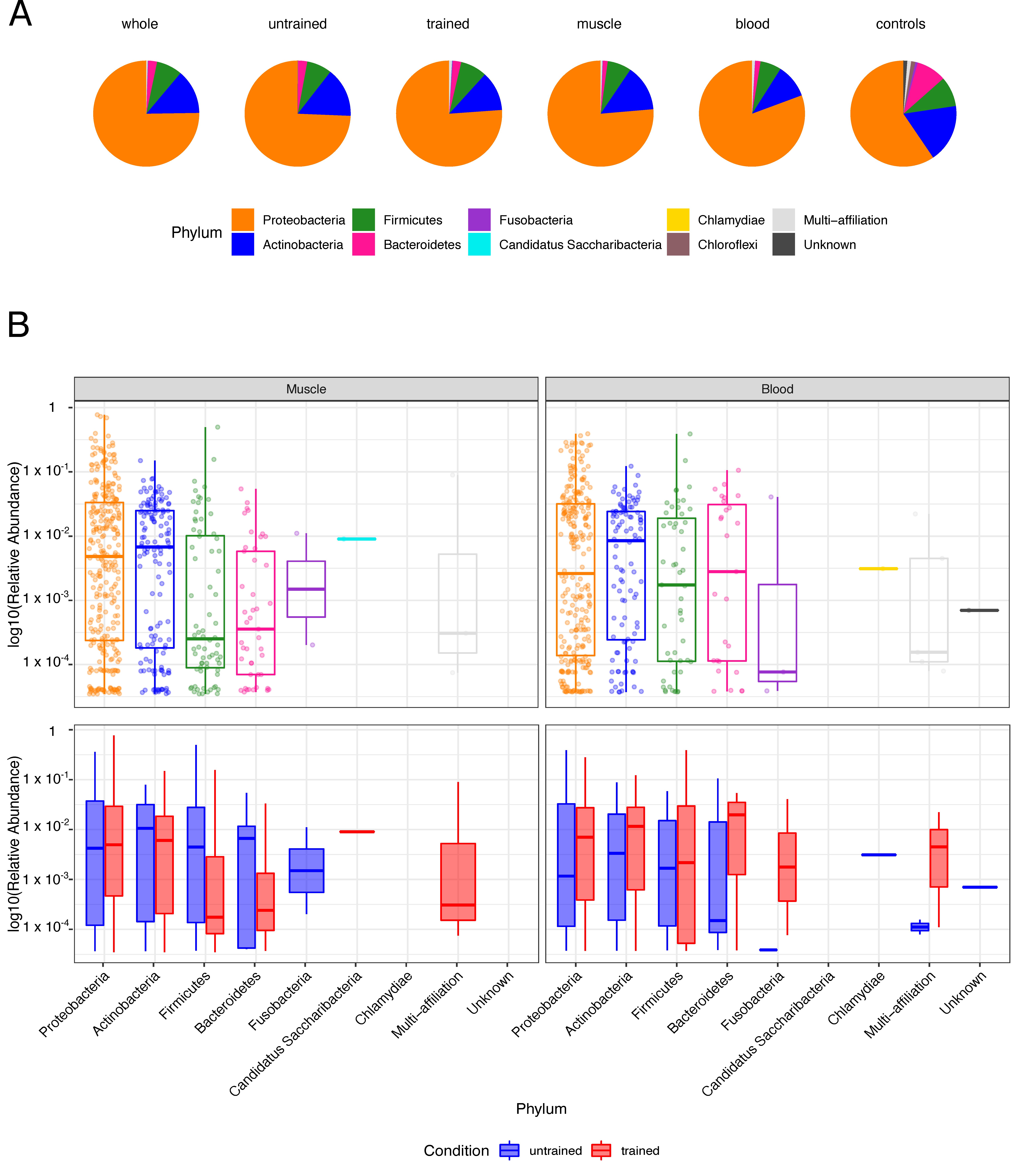
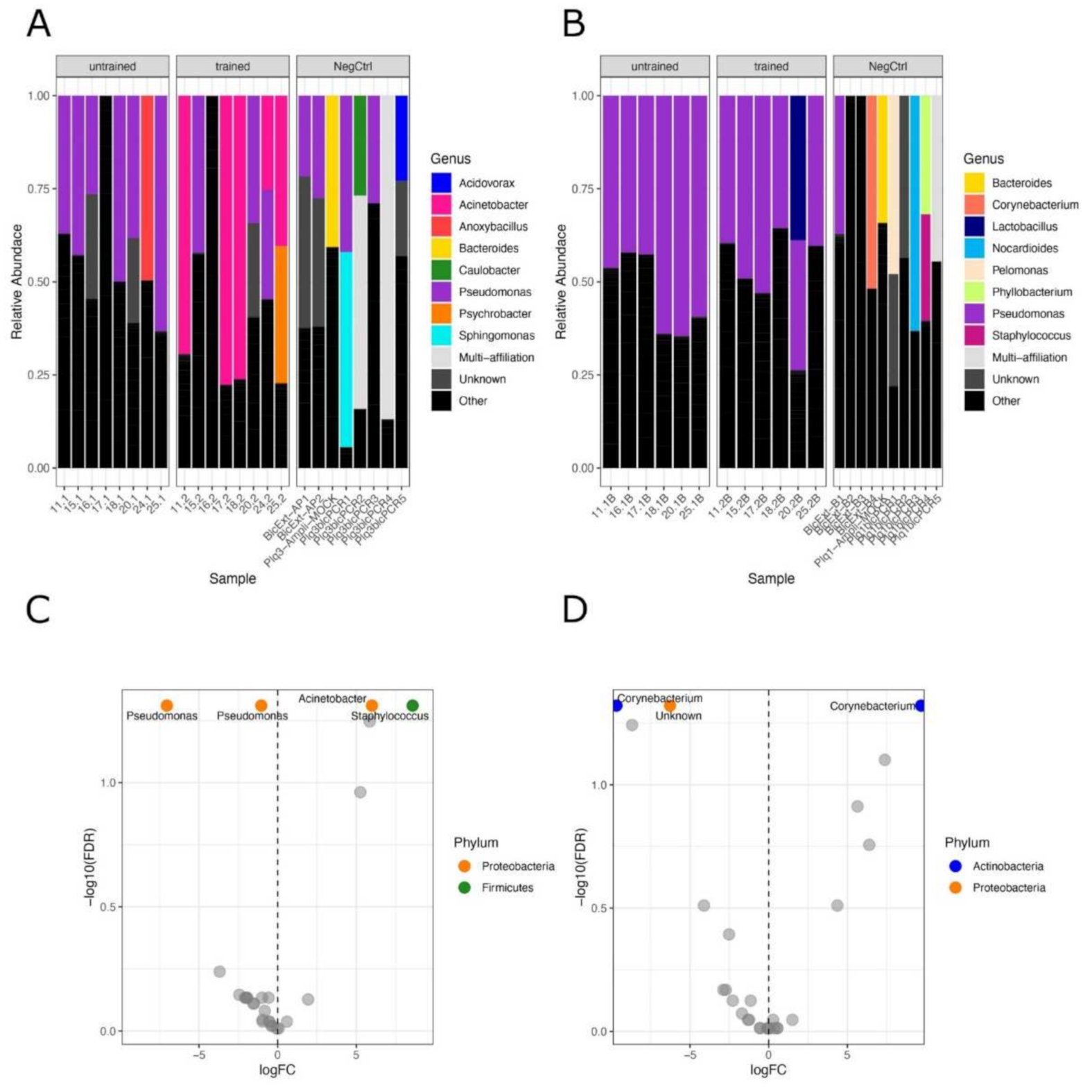


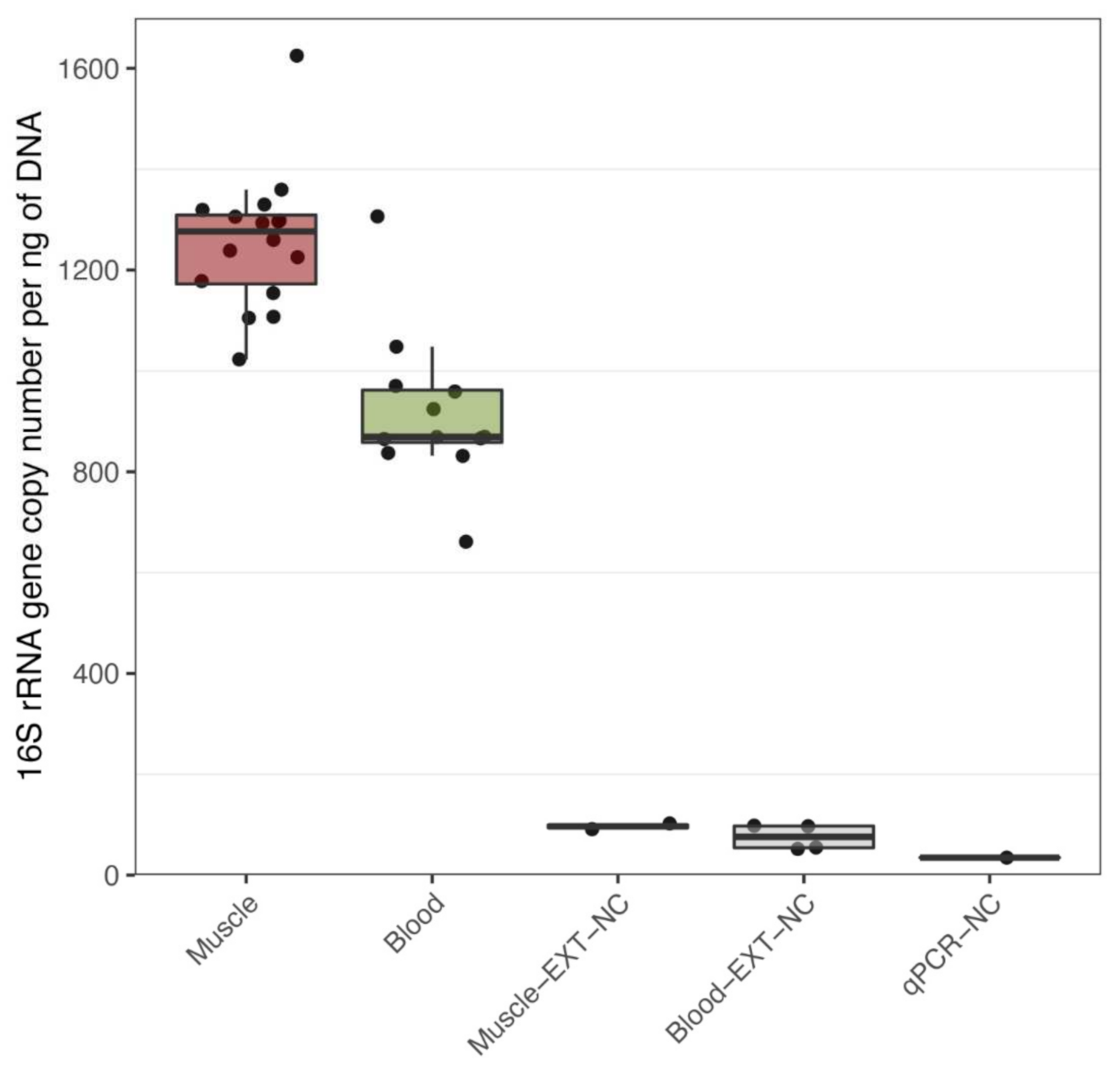{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Untrained, n = 8 | Trained, n = 8 | |
|---|---|---|
| Age—years | 24 ± 4 | 24 ± 4 |
| Weight—kg | 79.4 ± 10.0 | 78.5 ± 9.2 |
| Body mass index—kg/m2 | 22.89 ± 2.21 | 22.71 ± 2.08 |
| Waist—cm | 87 ± 7 | 80 ± 7 *** |
| Hip—cm | 94 ± 5 | 90 ± 5 ** |
| Waist/Hip | 0.93 ± 0.04 | 0.89 ± 0.05 * |
| VO2—mL | 3694 ± 514 | 4332 ± 458 *** |
| VO2/kg | 46.5 ± 4.4 | 55.4 ± 4.3 *** |
| Glucose (fasting)—mmol/L | 4.9 ± 0.4 | 5.1 ± 0.4 |
| Insulin—pmol/L | 64 ± 24 | 64 ± 35 |
| HOMA-IR | 2.31 ± 0.94 | 2.46 ± 1.56 |
| HbA1c—% | 34 ± 3 | 33 ± 3 |
| Plasma cholesterol (total)—mmol/L | 1.3 ± 0.8 | 4.4 ± 0.4 |
| Low-density lipoprotein—mmol/L | 6.7 ± 0.9 | 2.7 ± 0.4 |
| High-density lipoprotein—mmol/L | 1.2 ± 0.3 | 1.3 ± 0.2 * |
| Triglyceride—mmol/L | 4.6 ± 0.8 | 1.2 ± 0.3 |
| C-reactive protein—mg/L | 1.4 ± 0.2 | 1.0 ± 0.0 |
| Leukocytes—×109/L | 2.7 ± 0.6 | 6.1 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villarroel, J.; Donkin, I.; Champion, C.; Burcelin, R.; Barrès, R. Endurance Training in Humans Modulates the Bacterial DNA Signature of Skeletal Muscle. Biomedicines 2022, 10, 64. https://doi.org/10.3390/biomedicines10010064
Villarroel J, Donkin I, Champion C, Burcelin R, Barrès R. Endurance Training in Humans Modulates the Bacterial DNA Signature of Skeletal Muscle. Biomedicines. 2022; 10(1):64. https://doi.org/10.3390/biomedicines10010064
Chicago/Turabian StyleVillarroel, Julia, Ida Donkin, Camille Champion, Rémy Burcelin, and Romain Barrès. 2022. "Endurance Training in Humans Modulates the Bacterial DNA Signature of Skeletal Muscle" Biomedicines 10, no. 1: 64. https://doi.org/10.3390/biomedicines10010064
APA StyleVillarroel, J., Donkin, I., Champion, C., Burcelin, R., & Barrès, R. (2022). Endurance Training in Humans Modulates the Bacterial DNA Signature of Skeletal Muscle. Biomedicines, 10(1), 64. https://doi.org/10.3390/biomedicines10010064