
Zinc Homeostasis in Diabetes Mellitus and Vascular Complications


Abstract
:1. Introduction
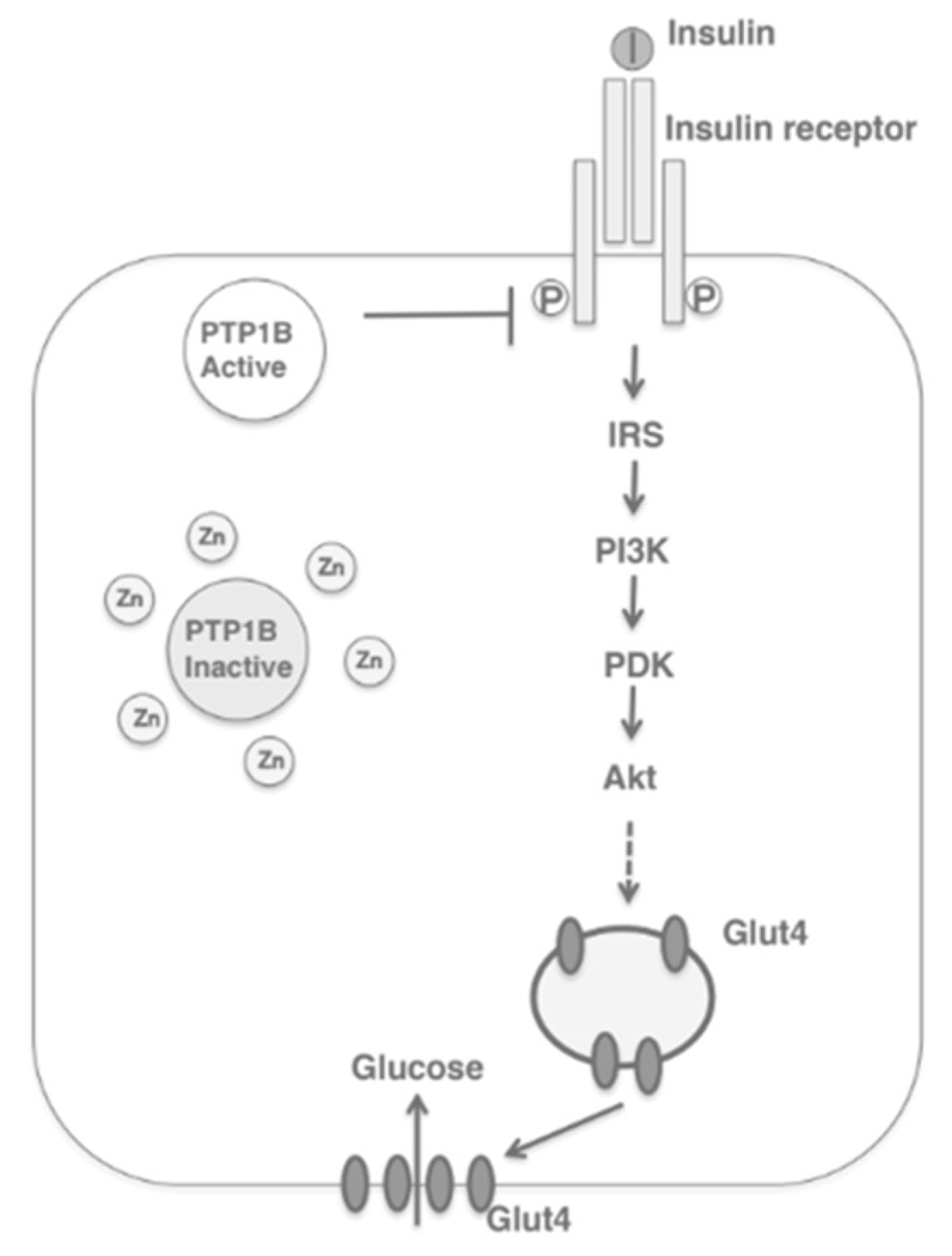
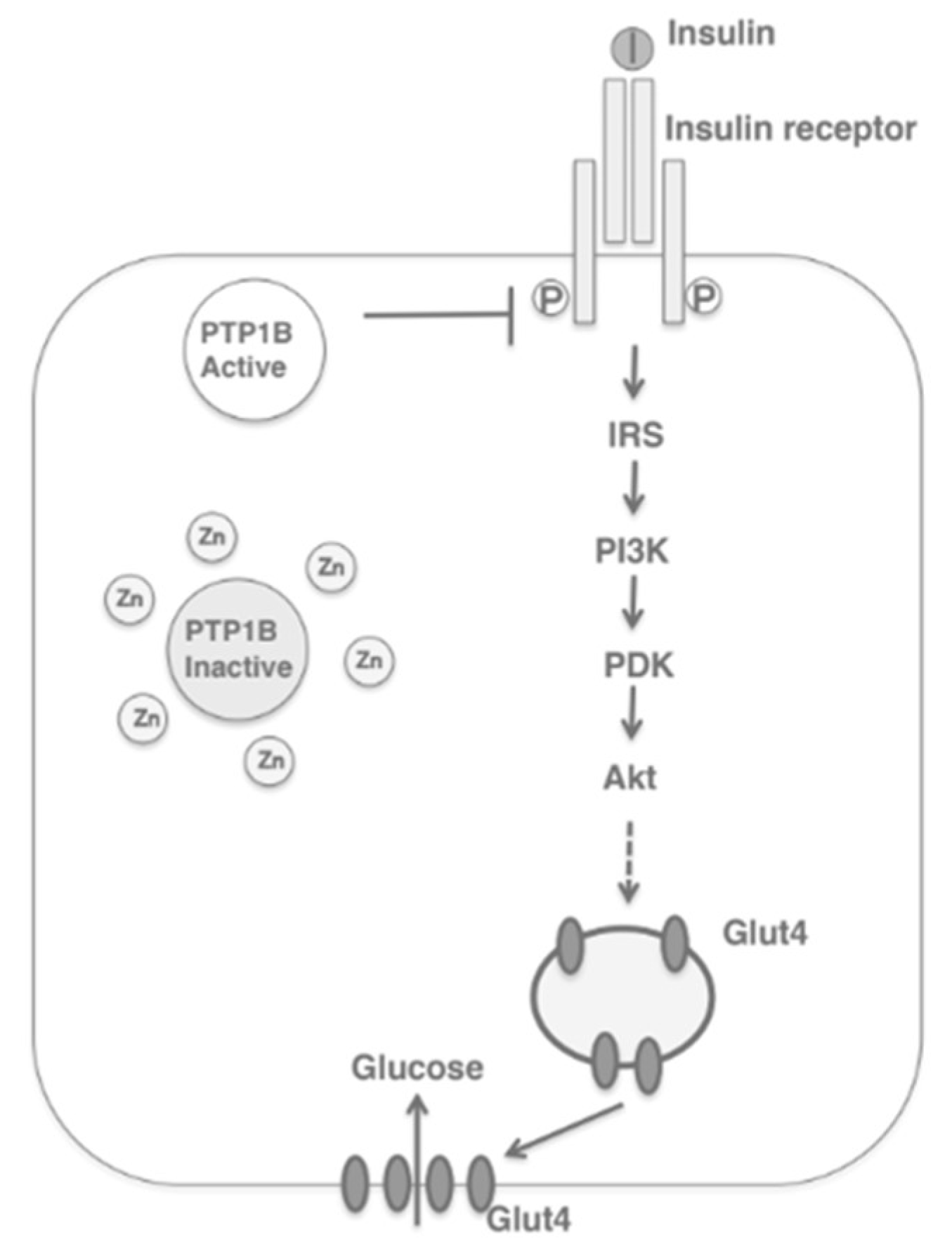
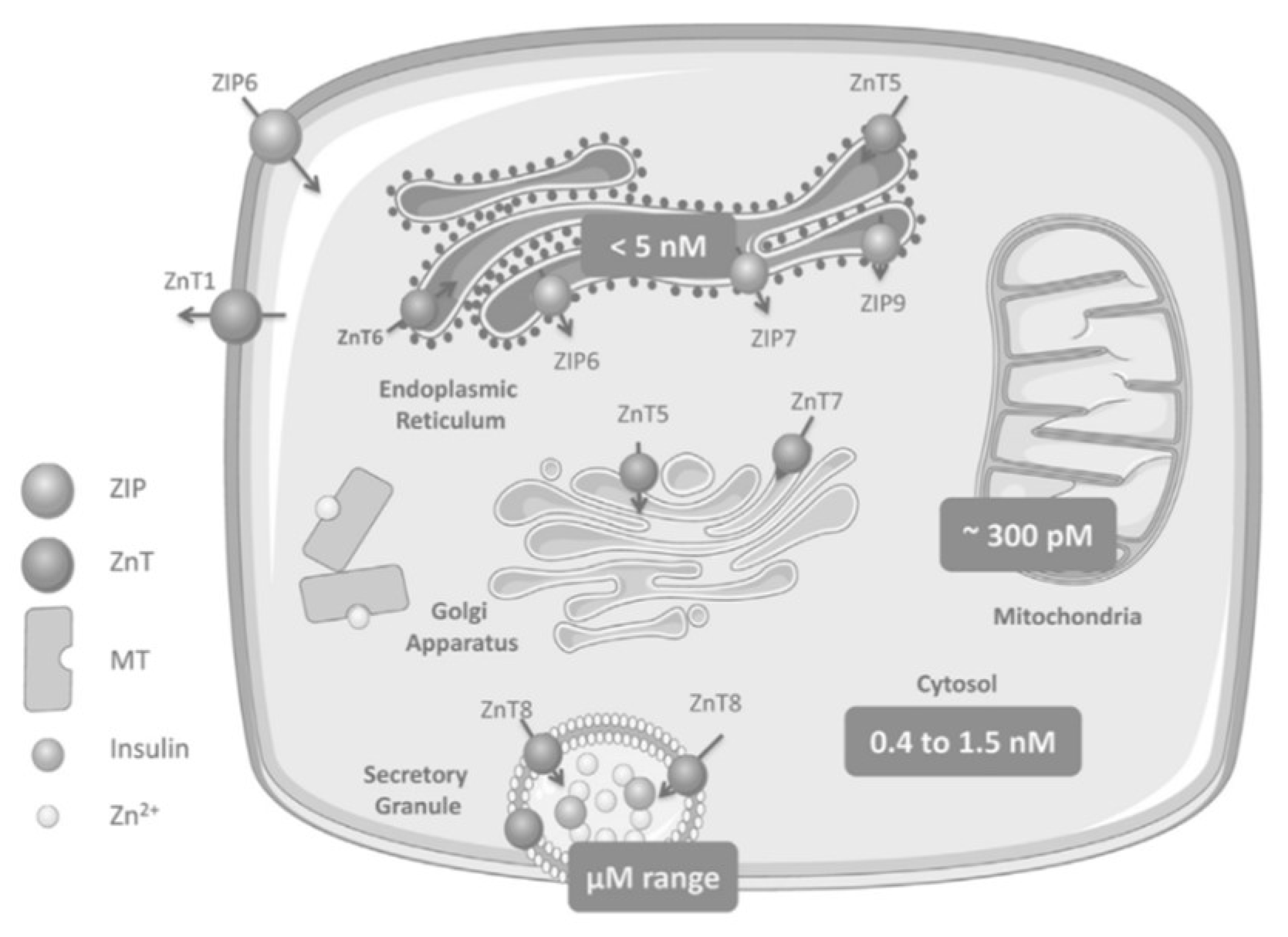
2. Zinc Homeostasis
Zinc Distribution in the β-Cell
3. Zinc Transporters: Glucose Homeostasis, Insulin Resistance and Immunity
3.1. ZnT8
3.2. ZIP7
3.3. Metallotionein
3.4. Other ZnT/SLC30A Transporters
3.5. Other ZIP/SLC39A Transporters
4. Zinc, Inflammation, Oxidative Stress and Vascular Complications
5. Zinc Supplementation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Cruz, K.J.C.; de Oliveira, A.R.S.; do Marreiro, D.N. Antioxidant role of zinc in diabetes mellitus. World J. Diabetes. 2015, 6, 333–337. [Google Scholar] [CrossRef]
- Do Marreiro, D.N.; Cruz, K.J.C.; Morais, J.B.S.; Beserra, J.B.; Severo, J.S.; de Oliveira, A.R.S. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Dai, S.-C.; Luan, X.; Chen, J.; Cannavicci, A. Dysfunction and Therapeutic Potential of Endothelial Progenitor Cells in Diabetes Mellitus. J. Clin. Med. Res. 2018, 10, 752–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Domingueti, C.P.; Dusse, L.M.S.; Carvalho, M.; Das, G.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Its Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Zhao, T.; Huang, Q.; Su, Y.; Sun, W.; Huang, Q.; Wei, W. Zinc and its regulators in pancreas. Inflammopharmacology 2019, 27, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Rutter, G.A. Zinc and diabetes. Arch Biochem Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Adulcikas, J.; Sonda, S.; Norouzi, S.; Sohal, S.S.; Myers, S. Targeting the Zinc Transporter ZIP7 in the Treatment of Insulin Resistance and Type 2 Diabetes. Nutrients 2019, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Norouzi, S.; Adulcikas, J.; Sohal, S.S.; Myers, S. Zinc transporters and insulin resistance: Therapeutic implications for type 2 diabetes and metabolic disease. J. Biomed. Sci. 2017, 24, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olechnowicz, J.; Tinkov, A.; Skalny, A.; Suliburska, J. Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J. Physiol. Sci. 2018, 68, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, M. Zinc Deficiency is Associated with Poor Glycemic Control. J. Coll. Physicians Surg. Pak. 2019, 29, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Fukunaka, A.; Fujitani, Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. Int. J. Mol. Sci. 2018, 19, 476. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.A.; Nield, A.; Myers, M. Zinc transporters, mechanisms of action and therapeutic utility: Implications for type 2 diabetes mellitus. J. Nutr. Metab. 2012, 2012, 173712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Batchuluun, B.; Ho, L.; Zhu, D.; Prentice, K.J.; Bhattacharjee, A.; Zhang, M.; Pourasgari, F.; Hardy, A.B.; Taylor, K.M.; et al. Characterization of Zinc Influx Transporters (ZIPs) in Pancreatic β Cells: Roles in regulating cytosolic zinc homeostasis and insulin secretion. J. Biol. Chem. 2015, 290, 18757–18769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Song, Y.; Elsherif, L.; Song, Z.; Zhou, G.; Prabhu, S.D.; Saari, J.T.; Cai, L. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation 2006, 113, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, B.; Wang, Y.; Tong, Q.; Liu, Q.; Sun, J.; Zheng, Y.; Cai, L. Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice. Int. J. Mol. Sci. 2017, 18, 580. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhao, J. The influence of zinc supplementation on metabolic status in gestational diabetes: A meta-analysis of randomized controlled studies. J. Matern.-Fetal Neonatal Med. 2021, 34, 2140–2145. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Wathurapatha, W.S.; Galappatthy, P.; Katulanda, P.; Jayawardena, R.; Constantine, G.R. Zinc supplementation in prediabetes: A randomized double-blind placebo-controlled clinical trial. J. Diabetes 2018, 10, 386–397. [Google Scholar] [CrossRef]
- Shan, Z.; Bao, W.; Zhang, Y.; Rong, Y.; Wang, X.; Jin, Y.; Song, Y.; Yao, P.; Sun, C.; Hu, F.B.; et al. Interactions between zinc transporter-8 gene (SLC30A8) and plasma zinc concentrations for impaired glucose regulation and type 2 diabetes. Diabetes 2014, 63, 1796–1803. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; van Dam, R.M.; Willett, W.C.; Hu, F.B. Prospective study of zinc intake and risk of type 2 diabetes in women. Diabetes Care 2009, 32, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, G.; Bouwkamp, C.G.; de Vrij, F.M.; Lovenberg, T.; Bonaventure, P.; Kushner, S.A.; Harrington, A.W. The Zinc Transporter SLC39A7 (ZIP7) Is Essential for Regulation of Cytosolic Zinc Levels. Mol. Pharmacol. 2018, 94, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, P.D.; Beltrame, J.F.; Wawer, A.A.; Abdo, A.I.; Murgia, C. Roles for endothelial zinc homeostasis in vascular physiology and coronary artery disease. Crit. Rev. Food Sci. Nutr. 2019, 59, 3511–3525. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Liu, X.; Pan, Z. Zinc deficiency and cellular oxidative stress: Prognostic implications in cardiovascular diseases. Acta Pharmacol. Sin. 2018, 39, 1120–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondragon, P.; Bergdahl, A. Metallothionein expression in slow- vs. fast-twitch muscle fibers following 4 weeks of streptozotocin-induced type 1 diabetes. Facets 2018, 3, 315–325. [Google Scholar] [CrossRef]
- Williams, C.L.; Long, A.E. What has zinc transporter 8 autoimmunity taught us about type 1 diabetes? Diabetologia 2019, 62, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Du, J.; Merriman, C.; Gong, Z. Genetic, Functional, and Immunological Study of ZnT8 in Diabetes. Int. J. Endocrinol. 2019, 2019, 1524905. [Google Scholar] [CrossRef]
- Anzilotti, C.; Swan, D.J.; Boisson, B.; Deobagkar-Lele, M.; Oliveira, C.; Chabosseau, P.; Engelhardt, K.R.; Xu, X.; Chen, R.; Alvarez, L.; et al. An essential role for the Zn2+ transporter ZIP7 in B cell development. Nat. Immunol. 2019, 20, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.A.; Nield, A.; Chew, G.-S.; Myers, M.A. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS ONE. 2013, 8, e79316. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose regulates free cytosolic Zn2+ concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet β-cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuncay, E.; Bitirim, C.V.; Olgar, Y.; Durak, A.; Rutter, G.A.; Turan, B. Zn2+-transporters ZIP7 and ZnT7 play important role in progression of cardiac dysfunction via affecting sarco(endo)plasmic reticulum-mitochondria coupling in hyperglycemic cardiomyocytes. Mitochondrion 2019, 44, 41–52. [Google Scholar] [CrossRef]
- Miao, X.; Wang, Y.; Sun, J.; Sun, W.; Tan, Y.; Cai, L.; Zheng, Y.; Su, G.; Liu, Q.; Wang, Y. Zinc protects against diabetes-induced pathogenic changes in the aorta: Roles of metallothionein and nuclear factor (erythroid-derived 2)-like 2. Cardiovasc. Diabetol. 2013, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Zhang, J.; Cai, L. Reappraisal of metallothionein: Clinical implications for patients with diabetes mellitus. J. Diabetes 2018, 10, 213–231. [Google Scholar] [CrossRef]
- Cai, L.U.; Wang, Y.; Zhou, G.; Chen, T.; Song, Y.; Li, X.; Kang, Y.J. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Li, Y.; Sun, X.; Sun, X. Antiapoptotic effect and inhibition of ischemia/reperfusion-induced myocardial injury in metallothionein-overexpressing transgenic mice. Am. J. Pathol. 2003, 163, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Chabosseau, P.; Woodier, J.; Cheung, R.; Rutter, G.A. Sensors for measuring subcellular zinc pools. Metallomics 2018, 10, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Cousins, R.J. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.S. Zinc is an Antioxidant and Anti-Inflammatory Agent: Its Role in Human Health. Front. Nutr. 2014, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Prasad, A.S.; Beck, F.W.; Fitzgerald, J.T.; Snell, D.; Bao, G.W.; Singh, T.; Cardozo, L.J. Zinc decreases C-reactive protein, lipid peroxidation, and inflammatory cytokines in elderly subjects: A potential implication of zinc as an atheroprotective agent. Am. J. Clin. Nutr. 2010, 91, 1634–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Tan, Y.; Dai, J.; Li, B.; Guo, L.; Cui, J.; Wang, G.; Shi, X.; Zhang, X.; Mellen, N.; et al. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol. Lett. 2011, 200, 100–106. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Li, H.; Wang, X.; Wu, W.; Gao, L. Effect and mechanisms of zinc supplementation in protecting against diabetic cardiomyopathy in a rat model of type 2 diabetes. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper-Capetini, V.; De Vasconcelos, D.A.A.; Martins, A.R.; Hirabara, S.M.; Donato, J., Jr.; Carpinelli, A.R.; Abdulkader, F. Zinc Supplementation Improves Glucose Homeostasis in High Fat-Fed Mice by Enhancing Pancreatic β-Cell Function. Nutrients 2017, 9, 1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Zhang, Q.; Sun, W.; Xin, Y.; Zhang, Z.; Tan, Y.; Zhou, S.; Zhang, C.; Cai, L.; Lu, X.; et al. Zinc treatment prevents type 1 diabetes-induced hepatic oxidative damage, endoplasmic reticulum stress, and cell death, and even prevents possible steatohepatitis in the OVE26 mouse model: Important role of metallothionein. Toxicol. Lett. 2015, 233, 114–124. [Google Scholar] [CrossRef]
- Barman, S.; Pradeep, S.R.; Srinivasan, K. Zinc supplementation alleviates the progression of diabetic nephropathy by inhibiting the overexpression of oxidative-stress-mediated molecular markers in streptozotocin-induced experimental rats. J. Nutr. Biochem. 2018, 54, 113–129. [Google Scholar] [CrossRef]
- Barman, S.; Pradeep, S.R.; Srinivasan, K. Zinc supplementation mitigates its dyshomeostasis in experimental diabetic rats by regulating the expression of zinc transporters and metallothionein. Metallomics 2017, 9, 1765–1777. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zinc Transporter | Regulators | Effect |
|---|---|---|
| ZnT1 | Metal-responsive mode of regulation; dietary intake of zinc | Efflux of zinc in smooth muscle cells |
| ZnT2 | Metal-responsive mode of regulation; dietary intake of zinc | Zinc transport in vesicles and lysosomes of pancreas, kidney, testis, epithelial cells, small intestine |
| ZnT3 | Glucose status | Transport of zinc to synaptic vesicles |
| ZnT4 | Unaffected by changes in dietary zinc uptake; regulated by extracellular zinc concentrations | Transport of zinc in the trans-Golgi network and in the cytoplasmic vesicular compartment |
| ZnT5 | Glucose status; zinc-responsive elements | Transport of zinc into Golgi lumen for storage |
| ZnT7 | Glucose status | Transport of zinc to Golgi apparatus in retina, liver, epithelial cells, small intestine; may play a redundant role of ZnT8 |
| ZnT8 | Glucose status | Regulation of zinc in the secretory vesicles of pancreatic β-cells |
| ZnT9 | Expressed in low levels in response to dietary intake of zinc | Export of zinc out of myocytes; efflux of zinc in smooth muscle cells |
| ZIP1 | Testosterone and prolactin | Uptake of zinc into cells |
| ZIP6 | Estrogen stimulation, glucose status | Down-regulation leads to poor insulin secretion |
| ZIP7 | Glucose status | Increases cytosolic zinc concentrations that participate in glucose mobilization and metabolism |
| ZIP8 | Glucose status, TNF-∝ in lung epithelial cells | Increases intracellular zinc levels |
| ZIP13 | Gene mutation leads to loss of function | Inhibition of adipocyte browning |
| ZIP14 | Acute phase response during inflammation; IL-6 | Rapid intake of plasma zinc into the organs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacKenzie, S.; Bergdahl, A. Zinc Homeostasis in Diabetes Mellitus and Vascular Complications. Biomedicines 2022, 10, 139. https://doi.org/10.3390/biomedicines10010139
MacKenzie S, Bergdahl A. Zinc Homeostasis in Diabetes Mellitus and Vascular Complications. Biomedicines. 2022; 10(1):139. https://doi.org/10.3390/biomedicines10010139
Chicago/Turabian StyleMacKenzie, Stephanie, and Andreas Bergdahl. 2022. "Zinc Homeostasis in Diabetes Mellitus and Vascular Complications" Biomedicines 10, no. 1: 139. https://doi.org/10.3390/biomedicines10010139
APA StyleMacKenzie, S., & Bergdahl, A. (2022). Zinc Homeostasis in Diabetes Mellitus and Vascular Complications. Biomedicines, 10(1), 139. https://doi.org/10.3390/biomedicines10010139