2. Experimental Section
2.1. General
Reagents and solvents were purchased from commercial suppliers and were used without purification unless otherwise specified. All experiments involving air- and/or moisture-sensitive compounds were carried out under N2 or Ar atmosphere. All reactions were monitored with analytical TLC (Merck Kieselgel 60 F254; Merck, Darmstadt, Germany). Column chromatography was carried out using FL-100D silica gel (Fuji Silysia, Aichi, Japan). Physical data were measured as follows. NMR spectra were recorded on JNM-ECS-300, JNM-ECS-400, and JNM-ECS-500 spectrometers (JEOL, Tokyo, Japan) using CDCl3 or DMSO-d6 as solvents with tetramethylsilane as an internal standard. IR spectra were recorded on a FT/IR-4200 spectrophotometer (JASCO, Tokyo, Japan). Optical rotations were recorded on a JASCO P-2200 instrument. FAB mass spectra were measured on a JEOL JMS-700 mass spectrometer. Solid-phase ON synthesis was performed on an nS-8 Oligonucleotide Synthesizer (GeneDesign, Osaka, Japan). MALDI-TOF mass spectra were recorded on an ultrafleXtreme mass spectrometer (Bruker Daltonics, Billerica, MA, USA) for oligonucleotides and on a JMS-S3000 (JEOL, Tokyo, Japan) for small molecules. Photo-irradiation experiments were conducted with a Xenon lamp (MAX-303; Asahi Spectra, Tokyo, Japan) using HQBP 450-VIS ø 25 and 365-VIS ø 25 as the optical filters. UV/Vis absorption measurements and UV melting experiments were performed using a UV-1650PC UV-Vis spectrophotometer equipped with a TMSPC-8 Tm analysis accessory (Shimadzu, Kyoto, Japan). ITC experiments were performed using a Microcal iTC200 (Malvern Instruments, Worcestershire, UK).
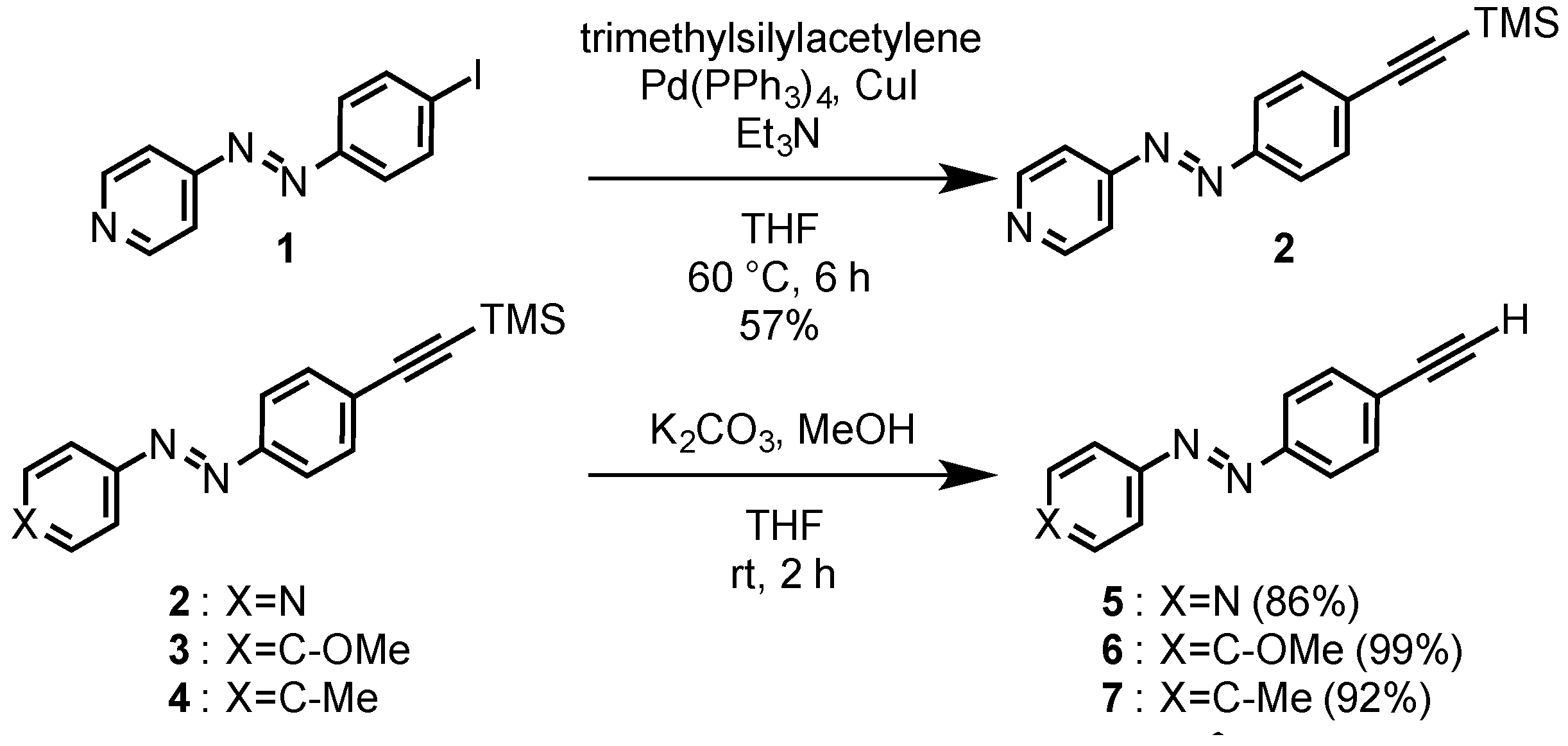
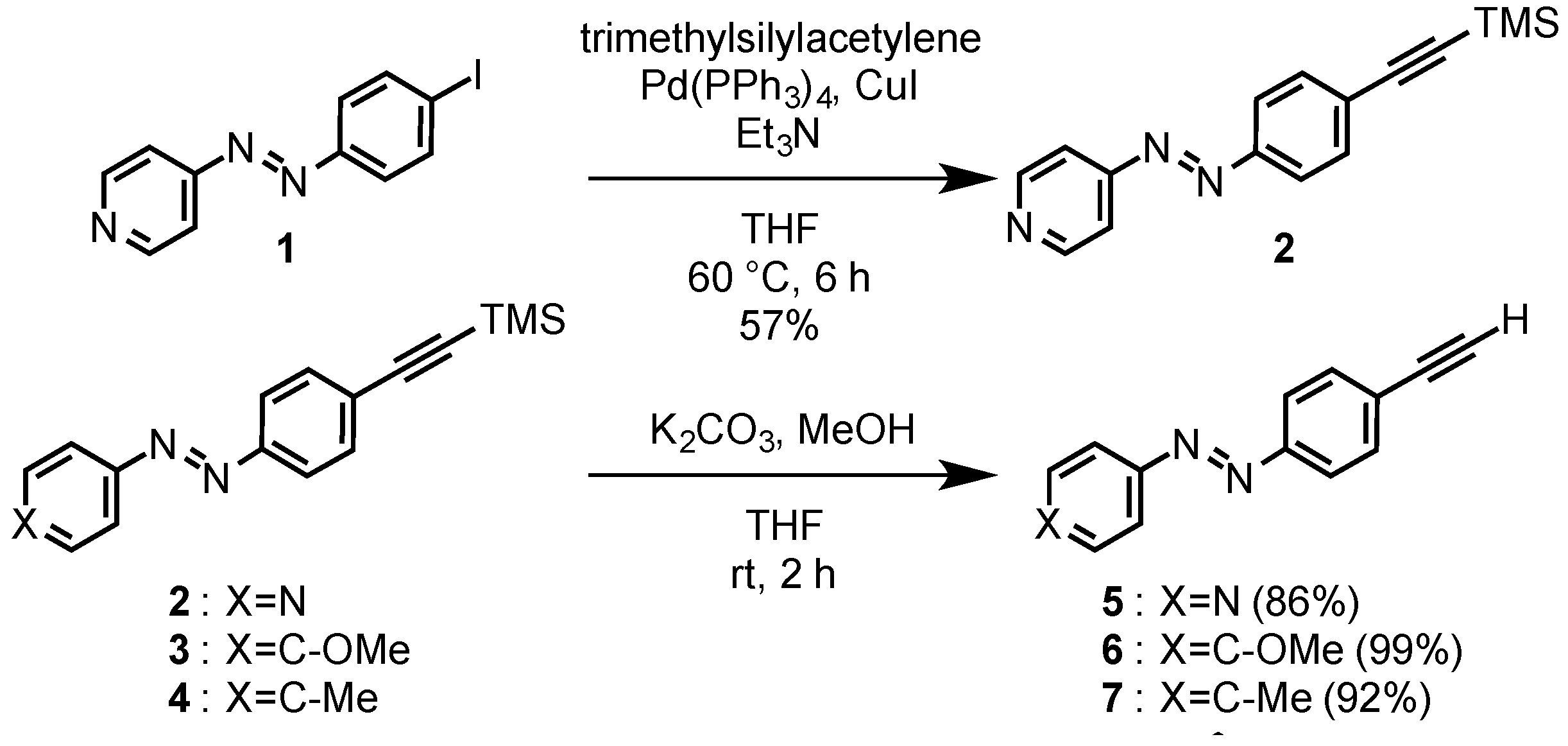
2.2. Preparation of 4-[4-(trimethylsilyl)ethynylphenylazo]pyridine (2)
Under an argon atmosphere, 4-(4-iodophenylazo)pyridine
1 [
18] (950 mg, 3.07 mmol) was dissolved in dry THF (30 mL). Pd(PPh
3)
4 (358 mg, 0.310 mmol), CuI (59.0 mg,0.310 mmol), Et
3N (2.1 mL, 15.4 mmol), and trimethylsilylacetylene (1.1 mL, 7.68 mmol) were then added. The reaction mixture was stirred at 60 °C for 6 h. The resultant mixture was filtered over Celite. The filtrate was concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane/AcOEt (8:1) to give compound
2 (485 mg, 57%) as a red oil. IR (KBr):
ν 2156 (C≡C), 1250 (N=N) cm
−1;
1H-NMR (400 MHz, CDCl
3):
δ 8.81 (2H, dd,
J = 2.0, 4.5 Hz), 7.91 (2H, dd,
J = 1.5, 6.5 Hz), 7.70 (2H, dd,
J = 1.5, 4.5 Hz), 7.63 (2H, dd,
J = 2.0, 6.5 Hz), 0.28 (9H, s);
13C-NMR (100 MHz, CDCl
3):
δ 151.5, 151.4, 133.0, 132.5, 130.4, 129.3, 123.5, 123.4, 116.33, 116.31; FAB-HRMS
m/z (MH
+) calcd for C
16H
18N
3Si: 280.1265; found 280.1272.
2.3. Preparation of 4-(4-ethynylphenylazo)pyridine (5)
Under an argon atmosphere, to a solution of compound
2 [
19] (319 mg, 1.14 mmol) in dry THF (5 mL) were added K
2CO
3 (79 mg, 0.57 mmol) and MeOH (5 mL) and the reaction mixture was stirred for 2 h at room temperature. The resultant mixture was diluted with AcOEt (30 mL) and washed with H
2O, followed by brine. The organic layer was dried (Na
2SO
4) and concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane/AcOEt (3:1) to give compound
5 (205 mg, 86%) as a red powder. M.p. 160 °C (decomposed); IR (KBr):
ν 2092 (C≡C), 1263 (N=N) cm
−1;
1H-NMR (400 MHz, CDCl
3):
δ 8.82 (2H, dd,
J = 1.5, 4.5 Hz), 7.92 (2H, d,
J = 8.0 Hz), 7.71 (2H, dd,
J = 1.5, 4.5 Hz), 7.66 (2H, d,
J = 9.0 Hz), 3.28 (1H, s);
13C-NMR (100 MHz, CDCl
3):
δ 157.0, 151.8, 151.4, 133.1, 126.2, 123.3, 116.2, 83.0, 80.3; FAB-HRMS
m/z (MH
+) calcd for C
13H
10N
3: 208.0869; found 208.0855.
2.4. Preparation of 4-ethynyl-4'-methoxyazobenzene (6)
Under an argon atmosphere, to a solution of compound
3 [
19] (750 mg, 2.44 mmol) in dry THF (10 mL) were added K
2CO
3 (169 mg, 1.22 mmol) and MeOH (10 mL) and the reaction mixture was stirred for 2 h at room temperature. The resultant mixture was diluted with AcOEt (40 mL) and washed with H
2O, followed by brine. The organic layer was dried (Na
2SO
4) and concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane/CH
2Cl
2 (4:1) to give compound
6 (576 mg, 99%) as an orange powder. M.p. 76–78 °C; IR (KBr):
ν 2102 (C≡C), 1252 (N=N) cm
−1;
1H-NMR (400 MHz, CDCl
3):
δ 7.81 (2H, d,
J = 8.5 Hz), 7.73 (2H, d,
J = 8.0 Hz), 7.51 (2H, d,
J = 8.0 Hz), 6.89 (2H, d,
J = 9.0 Hz), 3.76 (3H, s), 3.11 (1H, s);
13C-NMR (100 MHz, CDCl
3):
δ 162.3, 152.3, 146.9, 132.9, 124.9, 123.9, 122.4, 114.2, 83.4, 79.1, 55.5; FAB-HRMS
m/z (MH
+) calcd for C
15H
13N
2O: 237.1022; found 237.1021.
2.5. Preparation of 4-ethynyl-4'-methylazobenzene (7)
Under an argon atmosphere, to a solution of compound
4 [
19] (403 mg, 1.38 mmol) in dry THF (6 mL) were added K
2CO
3 (95 mg, 0.69 mmol) and MeOH (6 mL) and the reaction mixture was stirred for 2 h at room temperature. The resultant mixture was diluted with AcOEt (20 mL) and washed with H
2O, followed by brine. The organic layer was dried (Na
2SO
4) and concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane to give compound
7 (203 mg, 92%) as an orange powder. M.p. 138–140 °C; IR (KBr):
ν 2156 (C≡C), 1250 (N=N) cm
−1;
1H-NMR (300 MHz, CDCl
3):
δ 7.87–781 (4H, m), 7.61 (2H, d,
J = 8.5 Hz), 7.30 (2H, d,
J = 8.5 Hz), 3.21 (1H, s), 2.42 (3H, s);
13C-NMR (125 MHz, CDCl
3):
δ 152.2, 150.7, 142.0, 132.9, 129.8, 124.3, 123.0, 122.7, 83.3, 79.3, 21.5; MALDI-TOF-HRMS
m/z (MH
+) calcd for C
15H
13N
2: 221.1073; found 221.1075.
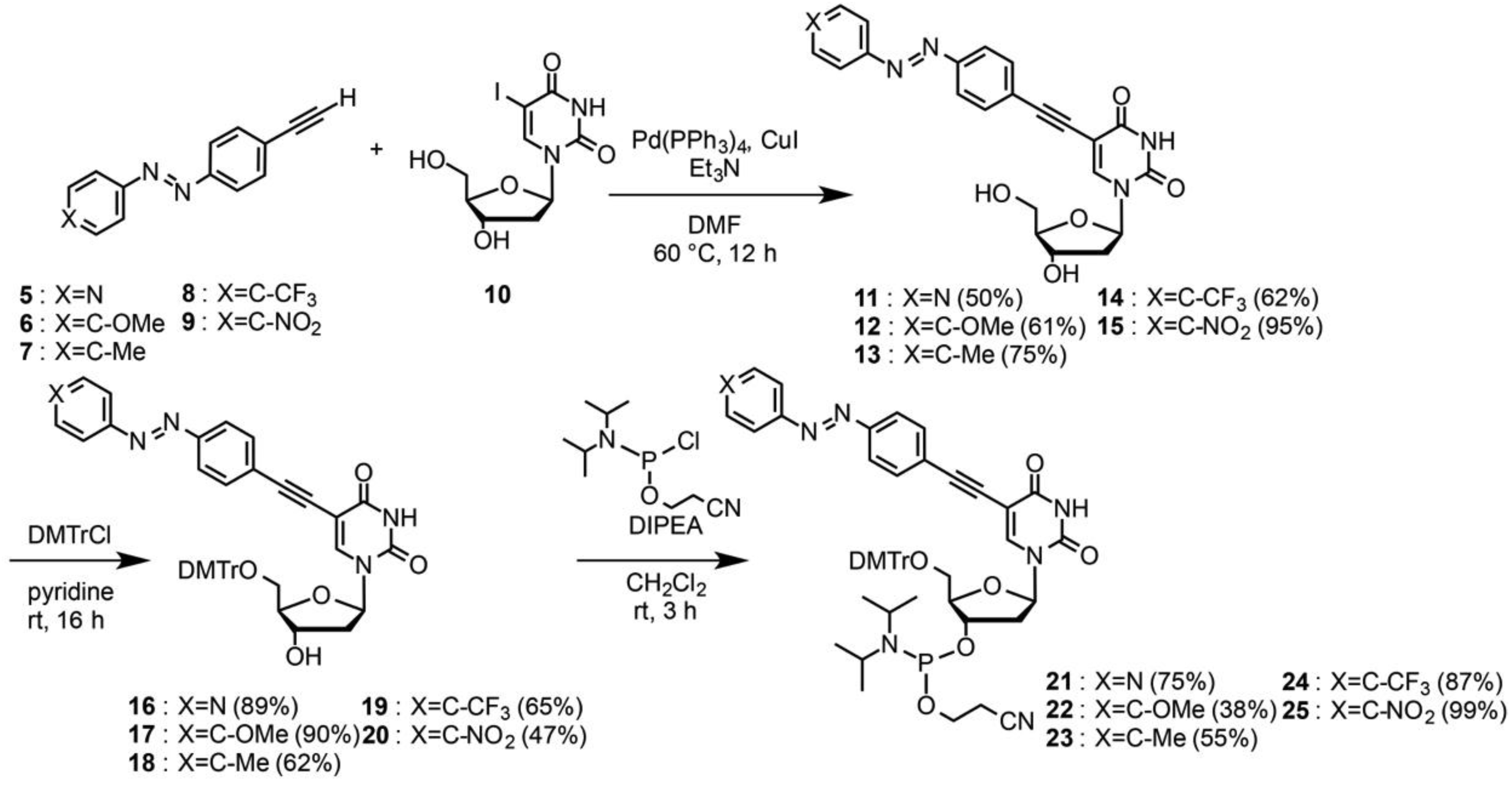
2.6. Preparation of 5-[4-(4-pyridyl)diazenylphenyl]ethynyl-2'-deoxyuridine (11)
Under an argon atmosphere, compound 5 (281 mg, 1.35 mmol) was dissolved in dry DMF (15 mL). Pd(PPh3)4 (156 mg, 0.135 mmol), CuI (26 mg, 0.135 mmol), Et3N (940 µL), and 2'-deoxy-5-iodouridine 10 (466 mg, 1.35 mmol) were then added. The reaction mixture was stirred at 60 °C for 12 h. The resultant mixture was filtered over Celite. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with DMF to give compound 11 (295 mg, 50%) as a red powder. M.p. 235 °C (decomposed); IR (KBr): ν 3250 (NH, OH), 1636 (C=O); [α]D24 −19.6 (c 1.00, DMSO); 1H-NMR (500 MHz, DMSO-d6): δ 11.7 (1H, br s, NH), 8.82 (2H, d, J = 4.5 Hz), 8.48 (1H, s, H-6), 7.96 (2H, d, J = 8.0 Hz), 7.75–7.68 (4H, m), 6.13 (1H, t, J = 6.0 Hz, H-1'), 5.27 (1H, d, J = 4.0 Hz, OH), 5.21 (1H, t, J = 4.5 Hz, OH), 4.29–4.25 (1H, m, H-3'), 3.84–3.82 (1H, m. H-4'), 3.67–3.58 (2H, m, H-5'), 2.21–2.17 (2H, m, H-2'); 13C-NMR (125 MHz, DMSO-d6): δ 161.3, 156.5, 151.5, 150.8, 149.4, 144.68, 132.3, 126.8, 123.5, 115.9, 97.7, 91.4, 87.6, 85.4, 85.0, 69.8, 60.8, 40.3; MALDI-TOF-HRMS m/z (MH+) calcd for C22H20N5O5: 434.1459; found 434.1451.
2.7. Preparation of 5-[4-(4-methoxyphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (12)
Under an argon atmosphere, compound 6 (303 mg, 1.28 mmol) was dissolved in dry DMF (15 mL). Pd(PPh3)4 (148 mg, 0.128 mmol), CuI (24 mg, 0.128 mmol), Et3N (1 mL), and 2'-deoxy-5-iodouridine 10 (441 mg, 1.28 mmol) were then added. The reaction mixture was stirred at 60 °C for 12 h. The resultant mixture was filtered over Celite. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (10:1) to give compound 12 (360 mg, 61%) as a light-orange powder. M.p. 253 °C (decomposed); IR (KBr): ν 3345 (NH, OH), 2102 (C≡C), 1638 (C=O) cm−1; [α]D24 −21.6 (c 1.00, DMSO); 1H-NMR (500 MHz, DMSO-d6): δ 11.7 (1H, br s, NH), 8.47–8.44 (1H, m, H-6), 7.90–7.85 (4H, m), 7.64 (2H, d, J = 6.0 Hz), 7.13 (2H, d, J = 5.0 Hz), 6.18–6.11 (1H, m, H-1'), 5.31–5.25 (1H, m,OH), 5.24–5.18 (1H, m, OH), 4.32–4.24 (1H, m, H-3'), 3.89–3.81 (4H, m, H-4', Ph-OMe), 3.70–3.59 (2H, m, H-5'), 2.23–2.14 (2H, m, H-2'); 13C-NMR (125 MHz, DMSO-d6): δ 162.3, 161.3, 151.2, 149.4, 146.2, 144.3, 132.2, 124.8, 122.6, 114.7, 97.9, 91.6, 87.6, 85.1, 84.9, 69.9, 60.8, 55.6, 40.3; FAB-HRMS m/z (MH+) calcd for C24H23N4O6: 463.1612; found 463.1613.
2.8. Preparation of 5-[4-(4-methylphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (13)
Under an argon atmosphere, compound 7 (280 mg, 1.27 mmol) was dissolved in dry DMF (13 mL). Pd(PPh3)4 (173 mg, 0.127 mmol), CuI (29 mg, 0.127 mmol), Et3N (1.33 mL), and 2’-deoxy-5-iodouridine 10 (292 mg, 0.846 mmol) were then added. The reaction mixture was stirred at 60 °C for 12 h. The resultant mixture was filtered over Celite. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (10:1) to give compound 13 (284 mg, 75%) as a light-orange powder. M.p. 255 °C (decomposed); IR (KBr): ν 3345 (NH, OH), 2091 (C≡C), 1641 (C=O) cm−1; [α]D24 −20.6 (c 1.00, DMSO); 1H-NMR (500 MHz, DMSO-d6): δ 11.7 (1H, br s, NH), 8.47–8.44 (1H, m, H-6), 7.92–7.79 (4H, m), 7.69–7.63 (2H, m), 7.43–7.38 (2H, m), 6.17–6.11 (1H, m, H-1'), 5.30–5.24 (1H, m, OH), 5.22–5.16 (1H, m, OH), 4.30–4.23 (1H, m, H-3'), 3.85–3.79 (1H, m, H-4'), 3.71–3.57 (2H, m, H-5'), 2.40 (3H, s), 2.22-2.13 (2H, m, H-2'); 13C-NMR (125 MHz, DMSO-d6): δ 161.3, 151.1, 150.1, 149.4, 144.4, 142.2, 132.2, 130.0, 125.1, 122.8, 97.7, 91.4, 87.6, 85.4, 85.0, 69.8, 60.8, 40.3, 21.1; MALDI-TOF-HRMS m/z (MH+) calcd for C24H23N4O5: 447.1663; found 447.1663.
2.9. Preparation of 5-[4-(4-trifluoromethylphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (14)
Under an argon atmosphere, compound
8 [
20] (369 mg, 1.35 mmol) was dissolved in dry DMF (15 mL). Pd(PPh
3)
4 (156 mg, 0.135 mmol), CuI (26 mg, 0.135 mmol), Et
3N (1 mL), and 2'-deoxy-5-iodouridine
10 (465 mg, 1.35 mmol) were then added. The reaction mixture was stirred at 60 °C for 12 h. The resultant mixture was filtered over Celite. The filtrate was concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl
3/MeOH (10:1) to give compound
14 (415 mg, 62%) as a light-orange powder. M.p. 255 °C (decomposed); IR (KBr):
ν 3365 (NH, OH), 1659 (C=O) cm
−1; [α]
D24 −16.7 (c 1.00, DMSO);
1H-NMR (500 MHz, DMSO-
d6):
δ 11.8 (1H, br s, NH), 8.50 (1H, s, H-6), 8.08 (2H, d,
J = 8.0 Hz), 7.99 (4H, d,
J = 8.5 Hz), 7.71 (2H, d,
J = 8.0 Hz), 6.15 (1H, t,
J = 6.5 Hz, H-1'), 5.29 (1H, d,
J = 3.5 Hz, OH), 5.24–5.21 (1H, m, OH), 4.29 (1H, s, H-3'), 3.84 (1H, d,
J = 3.5 Hz, H-4'), 3.71–3.61 (2H, m, H-5'), 2.22–2.18 (2H, m, H-2');
13C-NMR (125 MHz, DMSO-
d6):
δ 161.3, 154.0, 150.8, 149.4, 144.6, 132.3, 130.9, 126.8, 126.3, 123.4, 123.2, 97.7, 91.4, 87.6, 86.2, 85.0, 79.2, 69.8, 60.8, 40.3; MALDI-TOF-HRMS
m/z (MNa
+) calcd for C
24H
19N
4O
5F
3Na: 523.1200; found 523.1200.
2.10. Preparation of 5-[4-(4-nitrophenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (15)
Under an argon atmosphere, compound
9 [
21] (302 mg, 1.20 mmol) was dissolved in dry DMF (15 mL). Pd(PPh
3)
4 (139 mg, 0.120 mmol), CuI (23 mg, 0.120 mmol), Et
3N (1 mL), and 2'-deoxy-5-iodouridine
10 (414 mg, 1.20 mmol) were then added. The reaction mixture was stirred at 60 °C for 12 h. The resultant mixture was filtered over Celite. The filtrate was concentrated
in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl
3/MeOH (5:1) to give compound
15 (545 mg, 95%) as a red powder. M.p. 270 °C (decomposed); IR (KBr):
ν 3403 (NH, OH), 1631 (C=O) cm
−1; [α]
D24 −22.5 (c 1.00, DMSO);
1H-NMR (500 MHz, DMSO-
d6):
δ 11.8 (1H, br s, NH), 8.50–8.44 (3H, m), 8.10–8.08 (2H, m), 8.02–7.97 (2H, m), 7.74–7.68 (2H, m), 6.17–6.11 (1H, m, H-1'), 5.30–5.28 (1H, m, OH), 5.22 (1H, d,
J = 4.5 Hz, OH), 4.28 (1H, m, H-3'), 3.84 (1H, m, H-4'), 3.68–3.62 (2H, m, H-5'), 2.22–2.18 (2H, m, H-2');
13C-NMR (125 MHz, DMSO-
d6):
δ 161.3, 155.0, 150.9, 149.4, 148.6, 144.7, 132.3, 126.7, 125.1, 123.6, 123.5, 97.7, 91.5, 87.6, 86.5, 85.0, 69.8, 60.8, 40.3; MALDI-TOF-HRMS
m/z (MNa
+) calcd for C
23H
19N
5O
7Na: 500.1177; found 500.1169.
2.11. Preparation of 5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-pyridyl)diazenylphenyl]ethynyl-2'-deoxyuridine (16)
To a solution of compound 11 (147 mg, 0.339 mmol) in dry pyridine (4 mL) was added DMTrCl (138 mg, 0.406 mmol) at room temperature, and the reaction mixture was stirred for 16 h. The reaction was quenched by the addition of MeOH with 10 min stirring. The solvent was removed in vacuo, and the residue was partitioned between CHCl3 and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give compound 16 (223 mg, 89%) as a red foam. IR (KBr): ν 3323 (NH, OH), 1705 (C=O), 1252 (N=N) cm−1; [α]D24 −4.3 (c 1.00, DMSO); 1H-NMR (500 MHz, DMSO-d6): δ 11.7 (1H, br s, NH), 8.67 (2H, d, J = 4.5 Hz), 8.05 (1H, s, H-6), 7.68 (2H, d, J = 8.5 Hz), 7.59 (2H, d, J = 6.0 Hz), 7.28 (2H, d, J = 7.0 Hz), 7.17–7.09 (8H, m), 7.02–7.00 (1H, m), 6.70–6.68 (4H, m), 6.01 (1H, t, J = 6.5 Hz, H-1'), 5.23–5.21 (1H, m,OH), 4.19–4.17 (1H, m, H-3'), 3.82 (1H, s, H-4'), 3.49 (6H, s, OMe), 3.06–3.04 (2H, m, H-5'), 2.16–2.13 (2H, m, H-2'); 13C-NMR (125 MHz, DMSO-d6): δ 161.3, 158.1, 156.5, 151.6, 150.7, 149.3, 144.7, 143.7, 135.5, 135.4, 132.2, 129.6, 127.9, 127.6, 126.7, 126.6, 123.1, 115.9, 113.2, 98.1, 86.1, 86.0, 85.8, 85.3, 70.35, 63.5, 54.96, 40.4; MALDI-TOF-HRMS m/z (MH+) calcd for C43H38N5O7: 736.2766; found 736.2759.
2.12. Preparation of 5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-methoxyphenyl)diazenylphenyl] ethynyl-2'-deoxyuridine (17)
To a solution of compound 12 (101 mg, 0.218 mmol) in dry pyridine (3 mL) was added DMTrCl (89 mg, 0.262 mmol) at room temperature, and the reaction mixture was stirred for 16 h. The reaction was quenched by the addition of MeOH with 10 min stirring. The solvent was removed in vacuo, and the residue was partitioned between CHCl3 and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give compound 17 (152 mg, 90%) as an orange foam. IR (KBr): ν 3437, 3410 (NH, OH), 1701 (C=O), 1272 (N=N) cm−1; [α]D24 28.4 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 10.2 (1H, br s, NH), 8.28 (1H, s, H-6), 7.88 (2H, d, J = 7.0 Hz), 7.64 (2H, d, J = 7.0 Hz), 7.46 (2H, d, J = 7.0 Hz), 7.36 (4H, d, J = 7.5 Hz), 7.26–7.24 (3H, m), 7.11 (1H, d, J = 8.0 Hz), 6.81–6.74 (4H, m), 6.45–6.37 (1H, m, H-1'), 4.60–4.58 (1H, m, H-3'), 4.22–4.19 (1H, m, H-4'), 3.84 (3H, s), 3.49 (6H, s, OMe), 3.45 (1H, d, J = 10.0 Hz, H-5'), 3.31 (1H, d, J = 9.0 Hz, H-5'), 2.68–2.57 (1H, m, H-2'), 2.40–2.28 (1H, m, H-2'), 1.29–1.19 (1H, m, OH); 13C-NMR (125 MHz, CDCl3): δ 162.1, 161.8, 158.4, 151.6, 149.5, 146.9, 144.4, 135.5, 135.4, 132.3, 129.9, 129.8, 128.0, 127.8, 124.3, 122.1, 114.1, 113.2, 100.3, 93.5, 86.9, 86.0, 86.0, 82.2, 72.2, 60.3, 55.4, 55.0, 41.6; MALDI-TOF-HRMS m/z (MH+) calcd for C45H41N4O8: 765.2919; found 765.2912.
2.13. Preparation of 5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-methylphenyl)diazenylphenyl] ethynyl-2'-deoxyuridine (18)
To a solution of compound 13 (200 mg, 0.446 mmol) in dry pyridine (5 mL) was added DMTrCl (182 mg, 0.536 mmol) at room temperature, and the reaction mixture was stirred for 16 h. The reaction was quenched by the addition of MeOH with 10 min stirring. The solvent was removed in vacuo, and the residue was partitioned between CHCl3 and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane/AcOEt (1:2) to give compound 18 (208 mg, 62%) as an orange foam. IR (KBr): ν 3425 (NH, OH), 1707 (C=O), 1253 (N=N) cm−1; [α]D24 −27.9 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 9.09 (1H, br s, NH), 8.27 (1H, s, H-6), 7.80 (2H, d, J = 8.0 Hz), 7.67 (2H, d, J = 7.5 Hz), 7.45 (2H, d, J = 8.0 Hz), 7.35 (4H, d, J = 8.5 Hz), 7.31–7.24 (4H, m), 7.15 (1H, t, J = 7.0 Hz), 7.11 (2H, d, J = 8.5 Hz), 6.81–6.78 (4H, m), 6.38 (1H, t, J = 4.0 Hz, H-1'), 4.60–4.57 (1H, m, H-3'), 4.17–4.09 (1H, m, H-4'), 3.77–3.65 (6H, m, OMe), 3.50–3.46 (1H, m, H-5'), 3.40–3.28 (1H, m, H-5'), 2.67–2.52 (2H, m, H-2'), 1.86–1.82 (1H, m, OH); 13C-NMR (125 MHz, CDCl3): δ 161.2, 158.6, 151.7, 150.7, 149.2, 144.4, 141.9, 135.4, 132.4, 130.0, 129.9, 129.8, 128.1, 127.9, 127.1, 124.7, 122.9, 122.3, 113.4, 100.4, 93.6, 87.1, 86.8, 85.9, 72.4, 63.5, 55.2, 41.2, 21.5; MALDI-TOF-HRMS m/z (MNa+) calcd for C45H40N4O7Na: 771.2789; found 771.2780.
2.14. Preparation of 5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-trifluoromethylphenyl)diazenylphenyl] ethyn-yl-2'-deoxyuridine (19)
To a solution of compound 14 (114 mg, 0.228 mmol) in dry pyridine (3 mL) was added DMTrCl (93 mg, 0.273 mmol) at room temperature, and the reaction mixture was stirred for 16 h. The reaction was quenched by the addition of MeOH with 10 min stirring. The solvent was removed in vacuo, and the residue was partitioned between CHCl3 and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3 with 0.5% Et3N) to give compound 19 (118 mg, 65%) as an orange foam. IR (KBr): ν 3372 (NH, OH), 1669 (C=O), 1253 (N=N) cm−1; [α]D24 26.9 (c 1.00, CHCl3); 1H-NMR (300 MHz, CDCl3): δ 10.04 (1H, br s, NH), 8.25 (1H, s, H-6), 7.85 (2H, d, J = 8.5 Hz), 7.69–7.55 (4H, m), 7.41–7.12 (8H, m), 7.07–6.95 (3H, m), 6.71–6.69 (4H, m), 6.36–6.29 (1H, m, H-1'), 4.55–4.47 (1H, m, H-3'), 4.15–4.07 (1H, m, H-4'), 3.57 (6H, s, OMe), 3.42–3.34 (1H, m, H-5'), 3.25–3.18 (1H, m, H-5'), 2.60–2.42 (2H, m, H-2’), 2.32–2.21 (1H, m), 0.99–0.90 (1H, m, OH); 13C-NMR (75 MHz, CDCl3): δ 161.7, 158.5, 154.2, 151.2, 149.5, 135.5, 132.4, 129.88, 129.86, 128.0, 127.9, 127.0, 126.22, 126.18, 126.0, 125.9, 123.0, 122.7, 113.3, 100.2, 93.3, 87.0, 86.9, 86.1, 83.7, 83.1, 75.2, 72.3, 55.1, 24.7; MALDI-TOF-HRMS m/z (MNa+) calcd for C45H37N4O7F3Na: 825.2507; found 825.2497.
2.15. Preparation of 5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-nitrophenyl)diazenylphenyl] ethynyl-2'-deoxyuridine (20)
To a solution of compound 15 (150 mg, 0.314 mmol) in dry pyridine (3 mL) was added DMTrCl (128 mg, 0.377 mmol) at room temperature, and the reaction mixture was stirred for 16 h. The reaction was quenched by the addition of MeOH with 10 min stirring. The solvent was removed in vacuo, and the residue was partitioned between CHCl3 and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give compound 20 (115 mg, 47%) as a red foam. IR (KBr): ν 3452 (NH, OH), 1703 (C=O), 1251 (N=N) cm−1; [α]D24 −16.9 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 9.98 (1H, br s, NH), 8.38-8.22 (3H, m), 7.95 (2H, d, J = 7.0 Hz), 7.69 (2H, d, J = 7.0 Hz), 7.49-7.14 (8H, m), 7.06 (2H, d, J = 7.0 Hz), 6.83-6.72 (5H, m), 6.43-6.38 (1H, m, H-1'), 4.61-4.57 (1H, m, H-3'), 4.21-4.16 (1H, m, H-4'), 3.65 (6H, s, OMe), 3.50-3.45 (1H, m, H-5'), 3.33-3.26 (1H, m, H-5'), 2.66-2.59 (1H, m, H-2'), 2.34-2.27 (1H, m, H-2'), 1.28-1.17 (1H, m, OH); 13C-NMR (125 MHz, CDCl3): δ 161.6, 158.5, 155.5, 151.2, 148.6, 144.4, 135.5, 135.4, 132.4, 132.3, 129.9, 128.0, 127.9, 127.8, 127.0, 124.6, 124.5, 123.4, 123.3, 123.0, 122.9, 113.3, 113.2, 100.1, 87.0, 86.9, 72.2, 55.1; MALDI-TOF-HRMS m/z (MNa+) calcd for C44H37N5O9Na: 802.2483; found 802.2501.
2.16. Preparation of 3'-O-[2-cyanoethyl(diisopropylamino)phosphino]-5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-pyridyl)diazenylphenyl]ethynyl-2'-deoxyuridine (21)
Under an argon atmosphere, to a solution of compound 16 (223 mg, 0.312 mmol) in dry CH2Cl2 (3 mL) was added N,N-diisopropylamine (160 µL, 0.935 mmol) and 2-cyanoethyl-N,N'-diisopropylchlorophosphoramidite (105 µL, 0.468 mmol) at room temperature, and the reaction mixture was stirred for 3 h. The resultant mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give a 1:1 diastereomeric mixture of 21 (219 mg, 75%) as a red foam. IR (KBr): ν 3603 (NH), 1712 (C=O), 1251 (N=N) cm−1; [α]D24 32.3 (c 1.00, CHCl3); 1H-NMR (300 MHz, CDCl3): δ 8.84 (2H, m), 8.42 (0.3H, s, H-6), 8.37 (0.7H, s, H-6), 7.74–7.70 (4H, m), 7.51–7.50 (2H, m), 7.41–7.35 (4H, m), 7.31–7.16 (3H, m), 7.04 (2H, dd, J = 9.0, 8.5 Hz), 6.83–6.77 (4H, m), 6.42–6.36 (1H, m, H-1'), 4.72–4.61 (1H, m, H-3'), 4.27–4.19 (1H, m, H-5'), 3.93–3.74 (1H, m, H-5'), 3.71 (6H, s, OMe), 3.67–3.49 (4H, m, CH2CH2CN), 3.36–3.24 (1H, m, H-4'), 2.72–2.52 (2H, m, H-2'), 2.49–2.35 (2H, m), 1.28 (8.4 H, t, J = 7.0 Hz, ((CH3)2CH)2N), 1.17 (3.6H, t, J = 7.0 Hz, ((CH3)2CH)2N); 13C-NMR (125 MHz, CDCl3): δ 158.5, 157.1, 151.1, 144.33, 144.29, 132.8, 135.5, 135.4, 135.3, 132.4, 132.3, 129.9, 128.0, 127.9, 127.8, 127.0, 126.8, 122.91, 122.89, 117.5, 116.2, 113.2, 100.1, 93.0, 87.0, 85.8, 63.1, 58.4, 58.2, 58.1, 57.9, 55.1, 43.23, 43.17, 43.07, 43.01, 40.9, 24.6, 24.54, 24.48, 24.44, 24.39, 21.5, 20.4, 20.3, 20.2; 31P-NMR (120 MHz, CDCl3): δ 148.8, 148.4; MALDI-TOF-HRMS m/z (MNa+) calcd for C52H54N7O8NaP: 958.3664; found 958.3647.
2.17. Preparation of 3'-O-[2-cyanoethyl(diisopropylamino)phosphino]-5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-methoxyphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (22)
Under an argon atmosphere, to a solution of compound 17 (152 mg, 0.199 mmol) in dry CH2Cl2 (2 mL) was added N,N-diisopropylamine (103 µL, 0.600 mmol) and 2-cyanoethyl-N,N'-diisopropylchlorophosphoramidite (67 µL, 0.299 mmol) at room temperature, and the reaction mixture was stirred for 3 h. The resultant mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give a 3:1 diastereomeric mixture of 22 (72 mg, 38%) as an orange foam. IR (KBr): ν 3624 (NH), 1700 (C=O), 1253 (N=N) cm−1; [α]D24 19.4 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 9.76 (1H, br s, NH), 8.37–8.28 (1H, m, H-6), 7.89 (2H, d, J = 7.5 Hz), 7.66 (2H, t, J = 8.0 Hz), 7.51–7.47 (2H, m), 7.41–7.31 (4H, m), 7.31–7.25 (2H, m), 7.19–7.12 (1H, m), 7.08 (2H, dd, J = 6.0, 10.0 Hz), 7.01–6.96 (2H, m), 6.84–6.78 (4H, m), 6.41–6.31 (1H, m, H-1'), 4.70–4.62 (1H, m, H-3'), 4.31–4.29 (0.25H, m, H-4'), 4-18-416 (0.75H, m, H-4'), 3.87–3.82 (4H, m, CH2CH2CN), 3.82 (3H, s), 3.68 (6H, s, OMe), 3.64–3.48 (2H, m, H-5'), 2.76–2.58 (2H, m, H-2'), 2.47–2.35 (2H, m, ((CH3)2CH)2N), 1.28 (9H, t, J = 7.0 Hz, ((CH3)2CH)2N), 1.17 (3H, t, J = 7.0 Hz, ((CH3)2CH)2N); 13C-NMR (125 MHz, CDCl3): δ 162.0, 158.4, 151.6, 149.2, 146.8, 144.2, 144.2, 135.28, 135.25, 132.18, 132.17, 129.8, 129.78, 129.75, 127.9, 127.85, 127.82, 127.7, 126.9, 124.6, 124.3, 121.97, 121.95, 117.3, 114.1, 113.2, 100.2, 100.1, 93.3, 86.8 (d, J (C, P) = 3.5 Hz), 77.2, 77.0, 76.7, 63.0, 62.9, 58.2, 58.1, 55.4, 54.9, 44.9 (d, J (C, P) = 5.0 Hz), 24.4, 24.3, 22.8, 22.7, 20.2 (d, J (C, P) = 7.0 Hz); 31P-NMR (200 MHz, CDCl3): δ 149.5, 149.1; MALDI-TOF-HRMS m/z (MNa+) calcd for C54H57N6O9NaP: 987.3817; found 987.3826.
2.18. Preparation of 3'-O-[2-cyanoethyl(diisopropylamino)phosphino]-5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-methylphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (23)
Under an argon atmosphere, to a solution of compound 18 (170 mg, 0.227 mmol) in dry CH2Cl2 (3 mL) was added N,N-diisopropylamine (119 µL, 0.681 mmol) and 2-cyanoethyl-N,N'-diisopropylchlorophosphoramidite (78 µL, 0.341 mmol) at room temperature, and the reaction mixture was stirred for 3 h. The resultant mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with hexane/AcOEt (1:1) to give a 2:1 diastereomeric mixture of 23 (120 mg, 55%) as an orange foam. IR (KBr): ν 3610 (NH), 1699 (C=O), 1272 (N=N) cm−1; [α]D24 29.6 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 9.74 (1H, br s, NH), 8.35–8.26 (1H, m, H-6), 7.84–7.77 (2H, m), 7.71–7.63 (2H, m), 7.51–7.44 (2H, m), 7.41–7.33 (4H, m), 7.32–7.25 (5H, m), 7.12–7.03 (2H, m), 6.84–6.75 (4H, m), 6.41–6.31 (1H, m, H-1'), 4.72–4.60 (1H, m, H-3'), 4.29–4.19 (1H, m, H-4'), 3.93–3.74 (4H, m, CH2CH2CN), 3.70 (4H, s, OMe), 3.63 (2H, s, OMe), 3.37–3.12 (2H, m, H-5'), 2.72–2.50 (2H, m, H-2'), 2.50 (3H, s), 2.41–2.26 (2H, m, ((CH3)2CH)2N), 1.22–1.14 (8H, m, ((CH3)2CH)2N), 1.12–1.06 (4H, m, ((CH3)2CH)2N); 13C-NMR (125 MHz, CDCl3): δ 153.6, 146.7, 145.7, 139.3, 137.4, 136.8, 130.4, 127.4, 124.93, 124.91, 124.74, 124.72, 123.0, 122.9, 122.0, 121.9, 119.8, 117.9, 119.3, 112.5, 108.3, 88.5, 82.1, 80.9 (d, J (C, P) = 3.5 Hz), 68.8, 68.7, 58.2, 53.4, 53.3, 50.2, 48.4, 38.2, 35.9, 35.8, 19.5 (d, J (C, P) = 7.0 Hz), 16.5, 15.4, 15.3; 31P-NMR (200 MHz, CDCl3): δ 149.6, 149.2; MALDI-TOF-HRMS m/z (MNa+) calcd for C54H57N6O8NaP: 971.3868; found 971.3875.
2.19. Preparation of 3'-O-[2-cyanoethyl(diisopropylamino)phosphino]-5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-trifluoromethylphenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (24)
Under an argon atmosphere, to a solution of compound 19 (98 mg, 0.122 mmol) in dry CH2Cl2 (2 mL) was added N,N-diisopropylamine (63 µL, 0.366 mmol) and 2-cyanoethyl-N,N'-diisopropylchlorophosphoramidite (41 µL, 0.183 mmol) at room temperature, and the reaction mixture was stirred for 3 h. The resultant mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give a 1:1 diastereomeric mixture of 24 (106 mg, 87%) as an orange foam. IR (KBr): ν 3640 (NH), 1674 (C=O), 1251 (N=N) cm−1; [α]D24 32.3 (c 1.00, CHCl3); 1H-NMR (300 MHz, CDCl3): δ 8.39 (0.4H, s, H-6), 8.34 (0.6H, s, H-6), 8.01–7.95 (2H, m), 7.81–7.68 (4H, m), 7.51–7.45 (2H, m), 7.41–7.32 (4H, m), 7.21–7.13 (1H, m), 7.09–6.99 (2H, m), 6.85–6.75 (6H, m), 6.42–6.31 (1H, m, H-1'), 4.70–4.59 (1H, m, H-3'), 4.29–4.15 (1H, m, H-4'), 3.75–3.64 (4H, m, CH2CH2CN), 3.48 (6H, s, OMe), 3.43–3.18 (2H, m, H-5'), 2.70–2.60 (2H, m, H-2'), 2.50–2.33 (2H, m, ((CH3)2CH)2N), 1.21–1.14 (7.2H, m, ((CH3)2CH)2N), 1.10-1.04 (4.8H, m, ((CH3)2CH)2N); 13C-NMR (125 MHz, CDCl3): δ 161.2, 158.6, 154.3, 151.3, 149.1, 146.9, 144.4, 144.3, 142.7, 135.5, 135.4, 132.4, 132.3, 32.0, 129.97, 129.92, 129.7, 128.0, 127.9, 127.8, 127.0, 126.3, 126.2, 126.0, 125.9, 125.6, 123.0, 122.7, 120.3, 117.6, 117.4, 116.7, 113.3, 100.4, 100.2, 93.2, 87.2, 87.0, 86.3, 85.9, 85.8, 83.0, 77.4, 77.2, 77.0, 76.6, 75.0, 74.9, 73.7, 63.1, 58.4, 58.2, 58.1, 55.2, 55.4, 53.4, 50.7, 43.3, 43.2, 43.1, 43.0, 40.8, 24.8, 24.6, 24.56, 24.51, 24.47, 24.41, 22.8, 20.4, 20.3, 20.1; 31P-NMR (120 MHz, CDCl3): δ 148.9, 148.4; MALDI-TOF-HRMS m/z (MNa+) calcd for C54H54N6O8F3NaP: 1025.3585; found 1025.3596.
2.20. Preparation of 3'-O-[2-cyanoethyl(diisopropylamino)phosphino]-5'-O-(4,4'-dimethoxytrityl)-5-[4-(4-nitrophenyl)diazenylphenyl]ethynyl-2'-deoxyuridine (25)
Under an argon atmosphere, to a solution of compound 20 (111 mg, 0.142 mmol) in dry CH2Cl2 (2 mL) was added N,N-diisopropylamine (74 µL, 0.426 mmol) and 2-cyanoethyl-N,N’-diisopropylchlorophosphoramidite (48 µL, 0.213 mmol) at room temperature, and the reaction mixture was stirred for 3 h. The resultant mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel column chromatography and eluted with CHCl3/MeOH (20:1 with 0.5% Et3N) to give a 2:1 diastereomeric mixture of 25 (137 mg, 99%) as a red foam. IR (KBr): ν 3576 (NH), 1712 (C=O), 1252 (N=N) cm−1; [α]D24 −15.0 (c 1.00, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 9.45 (1H, br s, NH), 8.45–8.29 (3H, m), 8.04–7.99 (2H, m), 7.76–7.70 (2H, m), 7.53–7.47 (2H, m), 7.42–7.36 (4H, m), 7.31–7.26 (2H, m), 7.20–7.14 (1H, m), 7.11–7.03 (2H, m), 6.85–6.79 (4H, m), 6.42–6.35 (1H, m, H-1'), 4.71–4.63 (1H, m, H-4'), 4.30–4.25 (0.33H, m, H-3'), 4.24–4.20 (0.67H, m, H-3'), 3.89–3.75 (4H, m, CH2CH2CN), 3.72–3.69 (6H, m, OMe), 3.65–3.50 (2H, m, H-5'), 2.75–2.61 (2H, m, H-2'), 2.47–2.35 (2H, m, ((CH3)2CH)2N), 1.22–1.16 (8H, m, ((CH3)2CH)2N), 1.16–1.06 (4H, m, ((CH3)2CH)2N); 13C-NMR (125 MHz, CDCl3): δ 156.5, 153.3, 150.6, 146.2, 144.3, 143.7, 139.4, 137.8, 130.5, 127.5, 124.9, 123.0, 122.9, 122.0, 121.8, 119.7, 119.53, 119.52, 118.0, 112.6, 103.3, 93.2, 81.9 (d, J (C, P) = 3.5 Hz), 68.7, 68.4, 62.9, 62.8, 58.1, 58.0, 55.4, 54.9, 38.9 (d, J (C, P) = 5.0 Hz), 24.4, 24.3, 22.8, 22.7, 19.5 (d, J (C, P) = 7.0 Hz); 31P-NMR (200 MHz, CDCl3): δ 149.6, 149.3; MALDI-TOF-HRMS m/z (MNa+) calcd for C53H54N7O10NaP: 1002.3567; found 1002.3570.
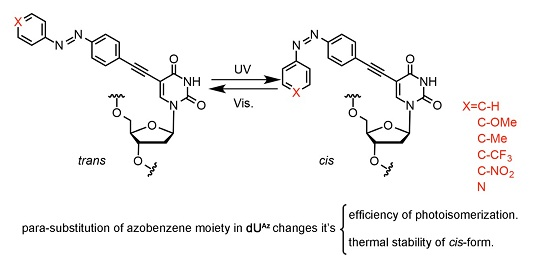
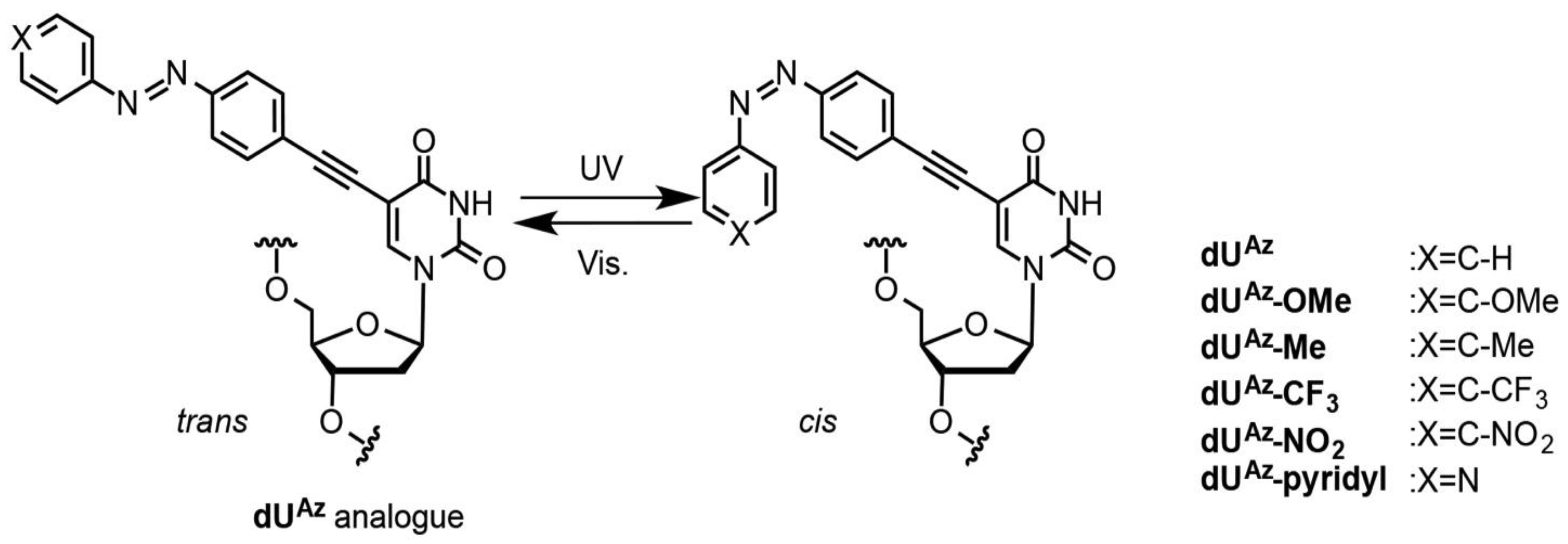
2.21. Synthesis of dUAz-Modified Oligodeoxynucleotides
Solid-phase oligonucleotide synthesis was performed using commercially available reagents and phosphoramidites with activator 42 (Sigma-Aldrich) as the activator.
para-Substituted-
dUAz phosphoramidites were chemically synthesized as described above. All of the reagents were assembled, and the ONs were synthesized according to the standard synthesis cycle (trityl on mode). Cleavage from the solid support and deprotection were accomplished with concentrated ammonium hydroxide solution at 55 °C for 12 h. The crude oligonucleotides were purified with Sep-Pak Plus C18 cartridges (Waters, MA, USA) followed by RP-HPLC on a XBridge
TM OST C18 column, 2.5 μm, 10 × 50 mm (Waters) using MeCN in 0.1 M triethylammonium acetate buffer (pH 7.0). The purified oligonucleotides were quantified by UV absorbance at 260 nm and confirmed by MALDI-TOF mass spectrometry (
Table 1).
Table 1.
Yields and MALDI-TOF MS data of dUAz-modified ONs.
Table 1.
Yields and MALDI-TOF MS data of dUAz-modified ONs.
| ON a | X | Yield/% | MALDI-TOF MS |
| Calcd. [M-H]− | Found [M-H]− |
| 28 | dUAz-OMe | 21 | 3852.6 | 3853.2 |
| 29 | dUAz-Me | 27 | 3836.6 | 3836.2 |
| 30 | dUAz-CF3 | 15 | 3890.6 | 3890.2 |
| 31 | dUAz-NO2 | 22 | 3866.6 | 3866.9 |
| 32 | dUAz-pyridyl | 21 | 3823.6 | 3824.2 |
2.22. UV Melting Experiments
Equimolecular amounts of the target DNA/RNA and ONs were dissolved in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl to give a final strand concentration of 4.0 µM. The melting samples were denatured at 100 °C and annealed slowly to room temperature. Absorbance was recorded in the forward and reverse directions at temperatures of 5–90 °C at a rate of 0.5 °C/min.
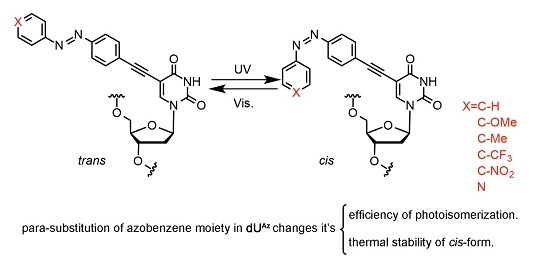
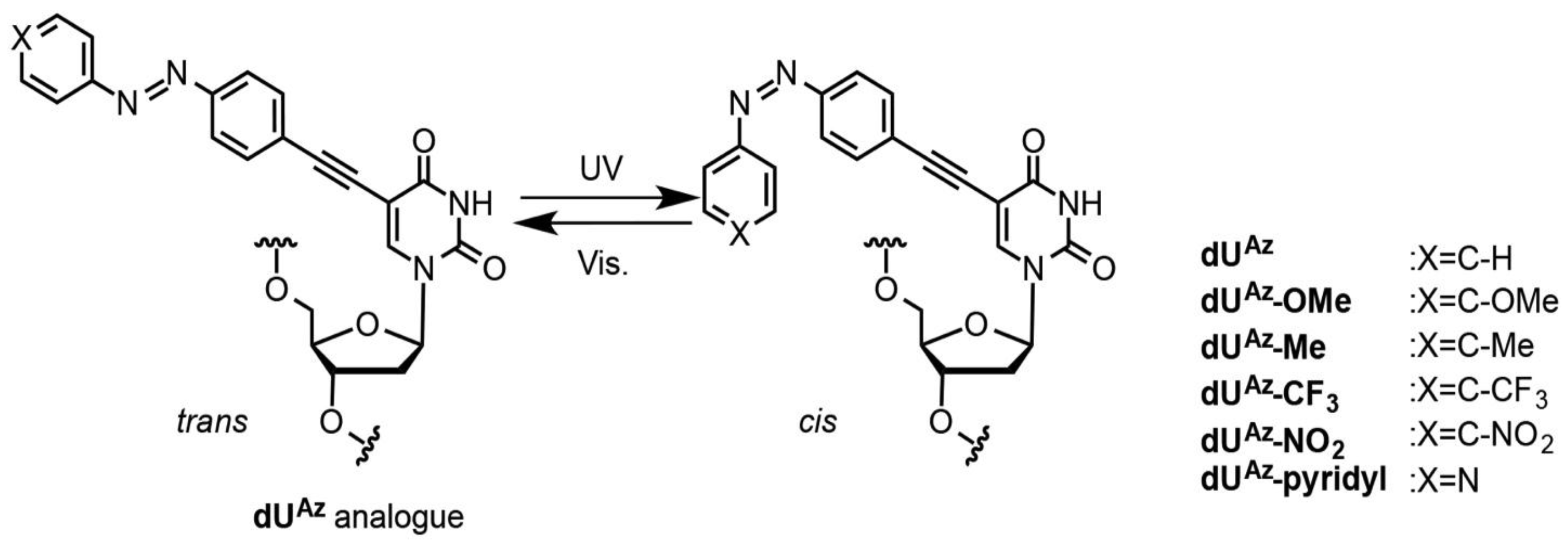
2.23. Photo-Isomerization of dUAz Analogues
ONs were dissolved in a 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl to give a final strand concentration of 4.0 µM. The ON solution was exposed to the appropriate wavelength of monochromic light for 10 seconds and subsequently analyzed by RP-HPLC. The ratio of cis/trans isomers was obtained from the HPLC peak areas at 260 nm.
2.24. Thermal Isomerization of cis-dUAz Analogues
ONs were dissolved in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl to give a final strand concentration of 4.0 µM. The oligonucleotide solution was exposed to the appropriate wavelength of monochromic light for 10 seconds and subsequently heated to 60 °C. The change of absorbance at 365 nm or 400 nm was monitored by a UV-Vis spectrophotometer and plotted to calculate the half-life time of cis-dUAz analogues.
2.25. Isothermal Titration Calorimetry
Prior to experiments, all solutions (titrant and titrand) were denatured at 100 °C and annealed slowly to room temperature. Titrations were carried out by injecting 2 µL portions of the complementary RNA strand into 250 µL of the dUAz-modified ON solution in the calorimetric cell. The dUAz-modified ON concentrations in the cell were 0.1 µM, and the solutions in the syringe were 1 µM. The equilibration period after each injection was 360 s. The measurements were performed with and without the photo-isomerization of dUAz-modified ONs.



{kind=link}
{kind=link}
{kind=link}
{kind=link}