1. Introduction
The rapid evolution of human lifestyles has led to significant environmental changes, particularly concerning air pollution, which poses a serious threat to both environmental and human well-being [
1,
2]. Among various air pollutants, hydrogen chloride (HCl) gas stands out due to its contribution to acid rain, generation of dioxins, and severe health effects, including respiratory issues and mucosal damage [
2,
3,
4,
5]. HCl’s origins in incineration plants, pharmaceutical, and metallurgical sectors, among others, necessitate effective monitoring to prevent environmental and health hazards [
3,
5]. Recent incidents of HCl vapor leaks in various locations globally underline the urgency of developing efficient detection methods [
6,
7]. Thus, the development of a highly sensitive platform for detecting hazardous HCl vapor becomes imperative to monitor and avert accidental threats. In recent years, several approaches like optochemical, optical, gas chromatographic, and electrochemical sensors have been developed for detecting HCl vapor, each with its limitations, such as time-consuming analysis, low sensitivity, and high cost [
8]. Presently, there is a growing demand for improved sensing materials capable of detecting HCl with remarkable sensitivity, focusing on materials like metal oxides, lanthanide complexes, and conducting molecules and aiming for lower detection limits [
9]. Among these, conducting materials draw significant attention due to their conductivity arising from conjugated π–π bonds, enabling their utilization in electrical and optical sensor devices [
9]. Organic π-conjugated small molecules offer distinct advantages, including customizable structures, mechanical flexibility, and solution processability, making them promising candidates for cost-effective, portable gas sensors operable at room temperature. However, challenges persist in fabricating organic semiconductor gas sensors, including issues related to sensitivity, recovery, and response time [
9,
10]. Over the years, various sensor technologies have been developed to detect gaseous HCl, including conductometric, amperometric, solid electrochemical, and optochemical sensors. However, these methods suffer from drawbacks, such as prolonged response times, limited sensitivity, high operational costs, and safety concerns. In light of these challenges, there is an urgent need for the development of more accessible, efficient, rapid, and safer sensors capable of onsite detection to effectively identify the presence of HCl.
In this context, Within the dynamic sphere of material science, covalent organic frameworks (COFs) have rapidly ascended as pivotal materials, especially in the cutting-edge field of sensor technology [
11,
12,
13]. These crystalline, porous structures that are ingeniously bonded by strong covalent connections between light elements, span a remarkable range of applications from gas storage and pollutant removal to catalysis, adsorption, and optoelectronics [
14]. Their deployment in the specific arena of chemical sensing, with a notable emphasis on HCl gas detection, exemplifies their expansive utility. Distinguished by their extensive surface areas, superior thermal resilience, and the capacity to form highly fluorescent frameworks, COFs transcend the traditional barriers faced by non-fluorescent variants. This advancement not only showcases their adaptable architectures and exceptional material qualities but also positions COFs at the forefront of research in material science, particularly as the exploration of their sensing capabilities continues to grow, heralding a new era of sensor development [
15,
16].
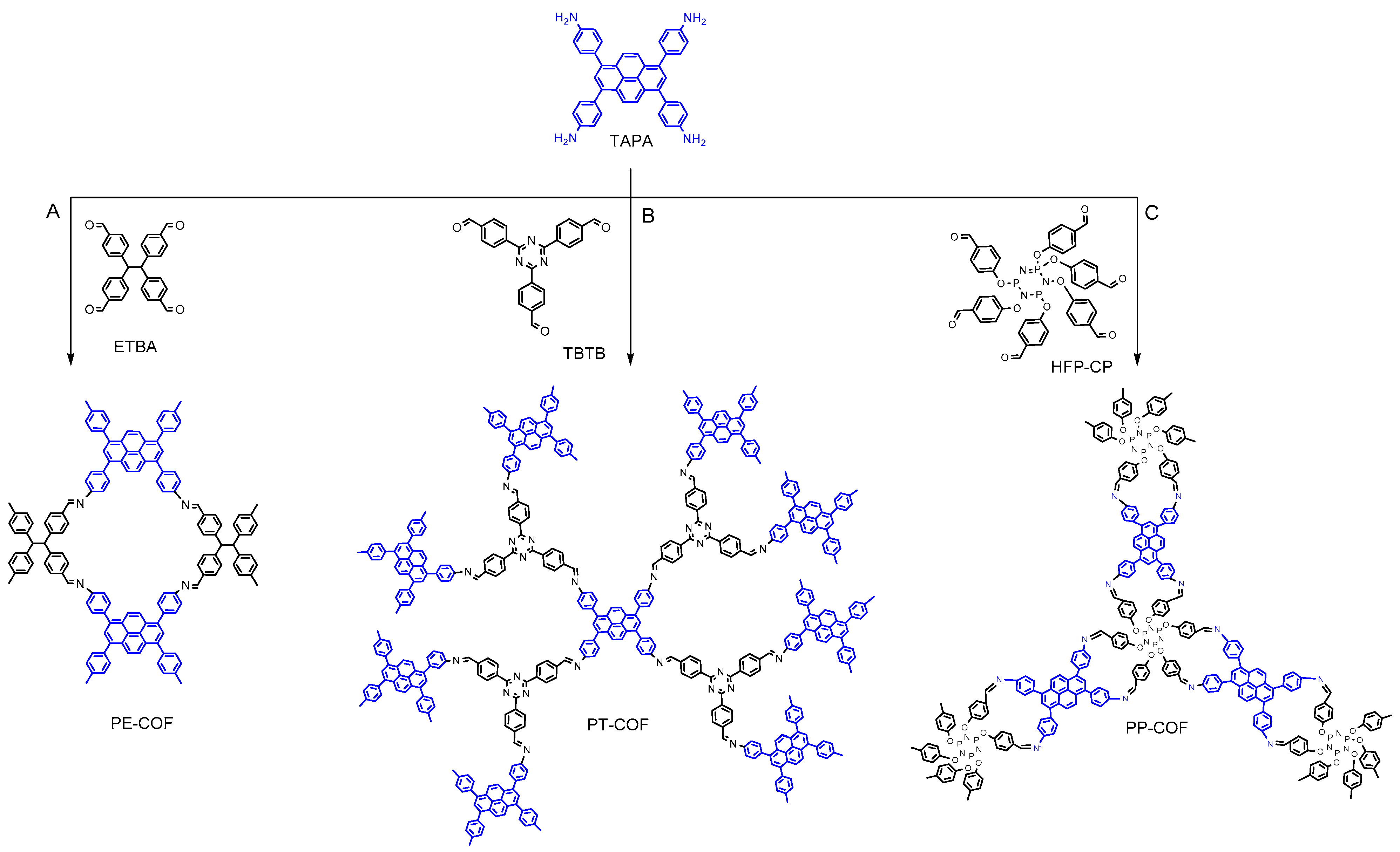
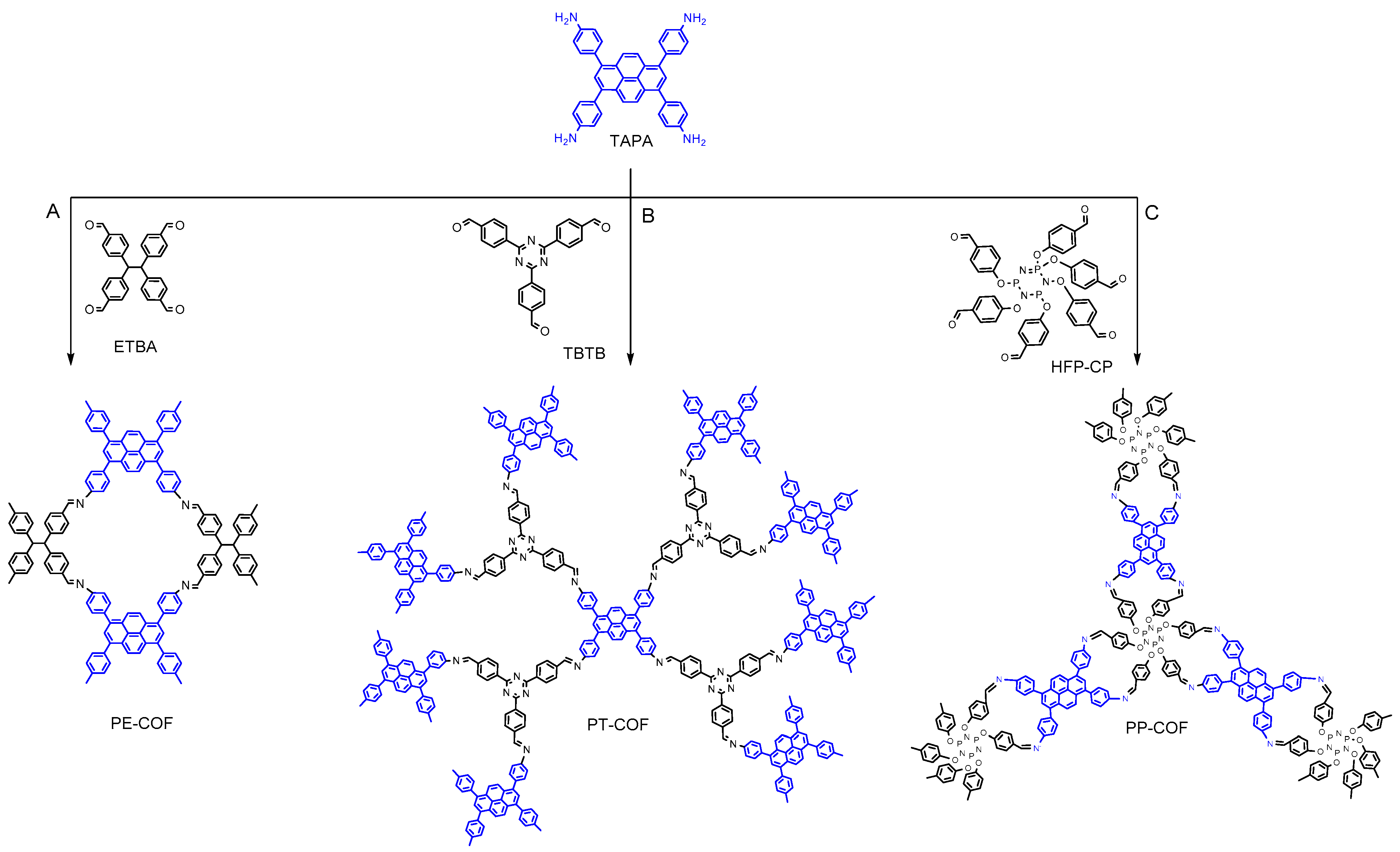
In this study, we synthesized three highly stable 2D COFs—PP–COF, PT–COF, and PE–COF—showcasing remarkable Brunauer–Emmett–Teller (BET) surface areas of up to 1350 m
2 g
−1. These COFs were created through Schiff base formation under solvothermal conditions, employing 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)tetraaniline and three distinct formyl species: 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde, hexakis (4-aldehyde phenoxy) cyclotriphosphazene, and 4,4′,4″,4‴-(ethane-1,1,2,2-tetrayl)tetra benzaldehyde, as illustrated in
Scheme 1. Capitalizing on their substantial surface areas, exceptional stability, and strong fluorescence, these COFs demonstrated their capacity as sensitive chemosensors for detecting HCl. Among the COFs studied, PE-COF initially emits a faint yellow fluorescence. However, upon exposure to solid-state HCl gas, it rapidly transforms into a vibrant dark orange emission in less than a second. This swift and notable color shift is easily observable to the naked eye under regular visible light. Our study aims to showcase an innovative system capable of detecting toxic acid vapors. Highlighting its ease of processing, cost-effectiveness, portability, and functionality under ambient conditions, we demonstrate its potential in constructing an electronic prototype for on-field applications. Our goal is to illustrate how this technology revolutionizes real-time monitoring of hazardous substances.
2. Results and Discussion
In this investigation, we capitalize on the distinctive electron configuration inherent in imine linkages to serve as the defining functional constituent within a series of two-dimensional covalent organic frameworks (COFs). These COFs were meticulously synthesized using 1,3,6,8-tetra(aminophenyl)pyrene (TAPA, as depicted in
Scheme 1) as a foundational building block. Upon subjecting the imine bonds within these materials to protonation, our observations reveal a noteworthy redshift in absorption, particularly in the near-infrared region, accompanied by the emergence of absorption bands induced by protonation. Importantly, these alterations manifest without compromising the structural integrity or crystalline attributes of the frameworks. We designed and created three COFs with outstanding stability and porosity using Schiff base reactions, as shown in
Scheme 1. In the synthesis of pyrene-based covalent organic frameworks (COFs), the approach centered on employing 1,3,6,8-tetra(aminophenyl)pyrene (TAPA) as a pivotal four-connected building block, serving as an amine linker. This strategic choice aimed to capitalize on TAPA’s structural versatility. The coupling of TAPA with diverse compounds, including 4,4′,4″,4‴-(ethane-1,1,2,2-tetrayl)tetra benzaldehyde (ETBA) (
Scheme 1A), 4,4′,4″-(1,3,5-Triazine-2,4,6-triyl)tribenzaldehyde (
Scheme 1B), and hexa(4-formyl-phenoxy)cyclotriphosphazene (
Scheme 1C). The synthesis process yielded three distinct two-dimensional (2D) structures termed PP–COF, PT–COF, and PE–COF. Detailed methods for synthesizing organic linkers, including reagents, reaction conditions, and purification techniques, are provided. The supporting data (
SI, Section S2, Schemes S1–S3 and Section S3, Figures S1–S6) encompass comprehensive characterization techniques, such as spectroscopic analysis (e.g., NMR). The creation of the three targeted COFs utilized a solvothermal technique [
17]. Acetic acid served as a catalyst, and the building blocks were suspended in a solution composed of a mixture of (mesitylene/1,4-dioxane) or o-(dichlorobenzene/n-butanol). Subsequently, the compounds were subjected to solvothermal conditions at 120 °C for 3–5 days (
SI, Section S4). The successful creation of imine bonds was shown by the FT-IR spectroscopy study of PP–COF, PT–COF, and PE–COF, which showed strong stretching vibrations of the C=N unit in the range of 1622–1625 cm
−1. Further evidence of the success of the Schiff base condensation process was provided by the spectra, which also revealed the removal of the N–H (3439–3398 cm
−1) stretching vibrations from the amino group in the amine linker and the C=O (1648–1660 cm
−1) vibration from the aldehyde linker (
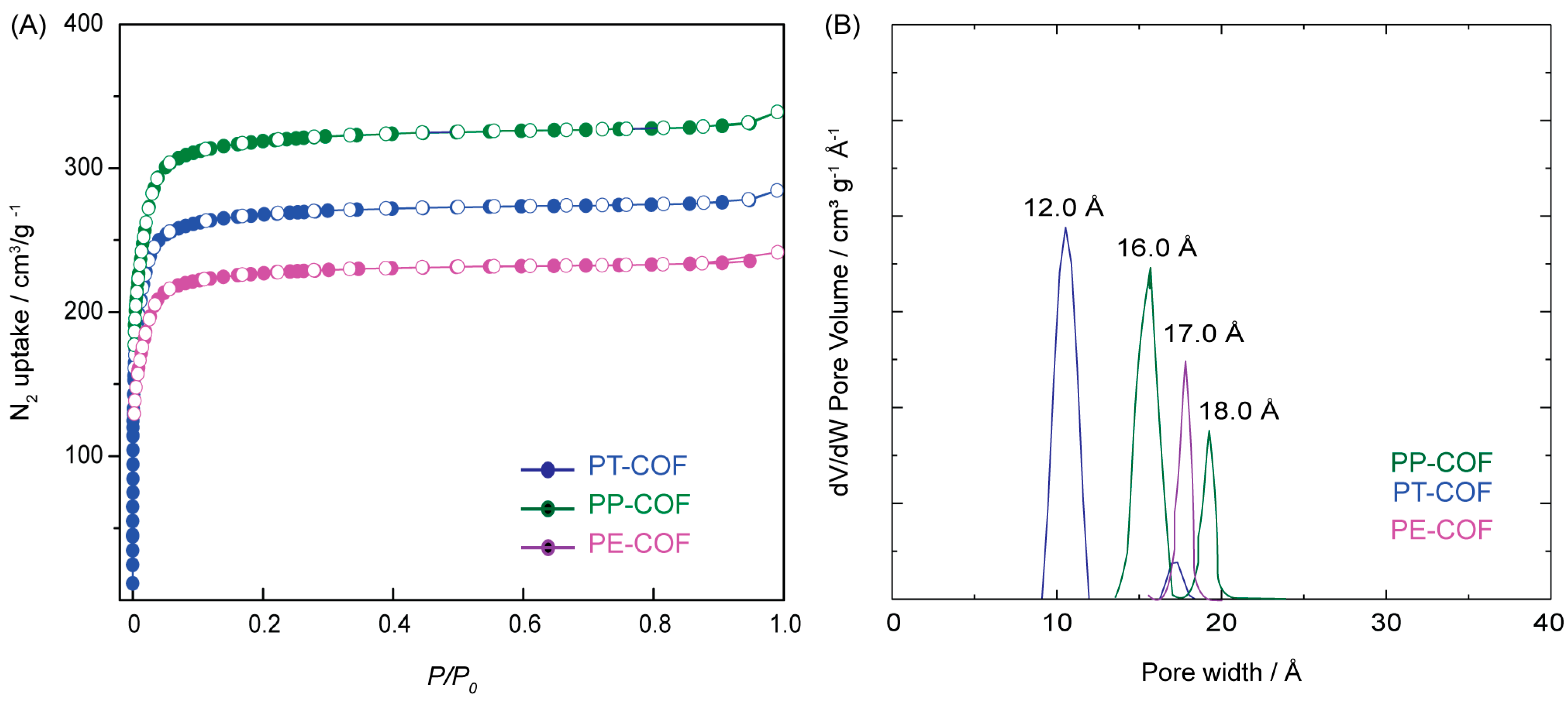
SI, Section S5, Figure S7). To examine the porous architectures of PP–COF, PT–COF, and PE–COF, nitrogen sorption isotherms at 77 K were employed (
Figure 1A). Prior to the nitrogen sorption measurement, the COF samples underwent an overnight pretreatment at 100 °C in a vacuum. The nitrogen absorption of PP–COF, PT–COF, and PE–COF displayed rapid increments at lower pressures (P/P
0 = 0 to 0.1), showcasing their microporous characteristics. The Brunauer–Emmett–Teller (BET) surface area measurements revealed values of 1350 m
2 g
−1, 1098 m
2 g
−1, and 730 m
2 g
−1 for PP–COF, PT–COF, and PE–COF, respectively. Evaluation of the total pore volumes at a P/P
0 ratio of 0.99 showcased a notable pore volume of 1.83 cm
3 g
−1 for PP–COF, surpassing both PT–COF (0.98 cm
3 g
−1) and PE–COF (1.23 cm
3 g
−1). Furthermore, nonlocal density functional theory (NLDFT) was employed to determine the pore sizes of the three COFs. The results revealed pore sizes of 16 Å and 18 Å for PP–COF and 12 Å and 17 Å for both PT–COF and PE–COF (
Figure 1B).
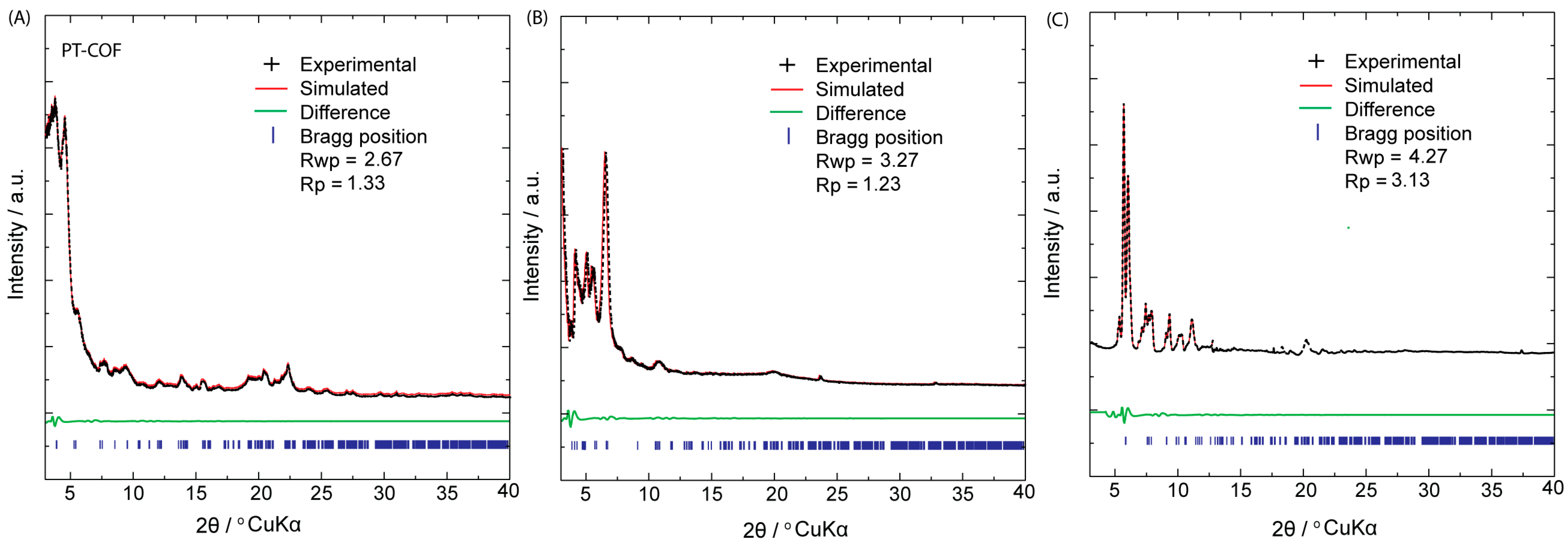
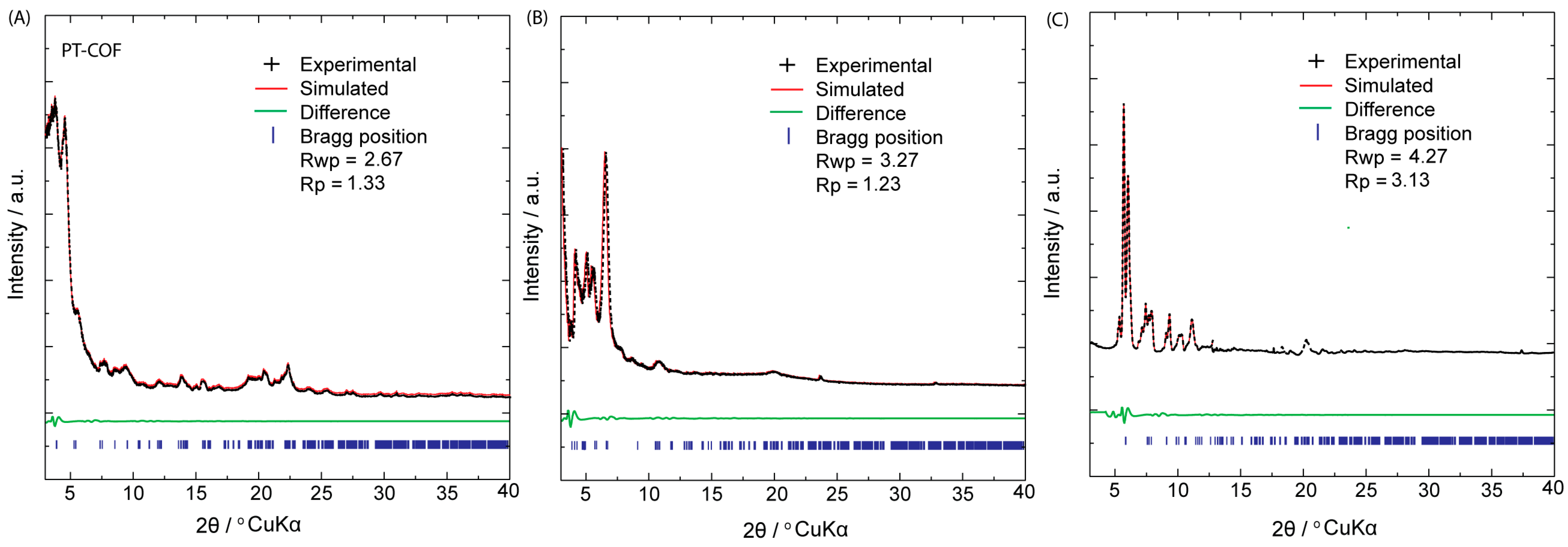
The crystalline characteristics of PP–COF, PT–COF, and PE–COF were investigated through powder X-ray diffraction (PXRD) analysis, as depicted in
Figure 2. In the PXRD diffractograms of PT–COF (
Figure 2A), distinct peaks corresponding to the (100), (110), (200), (210), (300), and (001) reticular planes were observed at 3.98°, 5.06°, 9.34°, 11.68°, 14.85°, and 24.80°, respectively. Similarly, the PXRD pattern of PP–COF (
Figure 2B) exhibited peaks at 2.68°, 5.58°, 9.82°, 12.54°, and 23.35°, which were attributed to the (110), (102), (200), (004), and (204) reticular planes, respectively. In the case of PE–COF (
Figure 2C), six distinctive peaks were identified, with the most intense peak at 5.56°, corresponding to the 100 facet, while others appeared at 6.78°, 7.31°, 9.52°, 11.67°, and 12.98°, representing the (110), (210), (220), (300), and (001) reticular planes, respectively. To gain comprehensive insights into their structures, the PXRD patterns of the investigated COFs underwent simulation via the Pawley refinement technique. The simulated PXRD patterns (depicted by the red curve in
Figure 2A–C) exhibited close alignment with the experimental PXRD patterns (depicted by the black curve in
Figure 2A–C). This agreement is evident from the difference pattern (indicated by the green curve in
Figure 2A–C), which confirms the accuracy of the structural analysis. In the case of PT–COF (
Figure 2A), the experimental PXRD pattern exhibited strong conformity with the simulated patterns derived from the AA-staggered stacking model, displaying favorable agreement factors (Rp = 1.33% and Rwp = 2.67%) with optimized parameters (a = b = 19.46 Å, c = 3.64 Å, α = β = 90°, and γ = 120°, resulting in a unit cell volume of 1279.51 Å
3). For PP–COF, the cell parameters were finetuned through Pawley refinements, showcasing minimal distinction between the experimental curve and simulated profiles produced by the AA-staggered stacking model (a = b = 30.17 Å, c = 3.43 Å, α = β = 90°, and γ = 120°, leading to a unit cell volume of 2704.10 Å
3), yielding negligible residuals (Rp = 1.23% and Rwp = 3.27%) (
Figure 2B). Moreover, the experimental PXRD patterns of PE–COF (
Figure 2C) strongly aligned with the XRD pattern generated from the AA-eclipsed stacking models, indicating excellent agreement. Conversely, significant disparities were observed when comparing the pattern derived from their corresponding AB’ staggered stacking model with the experimental results. The refined unit cell parameters for PE–COF were determined as a = 21.23 Å, b = 21.20 Å, c = 4.37 Å, with α = β = γ = 90°, revealing residuals of Rwp = 3.13% and Rp = 4.27% and a unit cell volume of 1964.51 Å
3 (
SI, Section S5, Figure S8) and (
SI, Section S10, Tables S1–S3).
In our investigation, we aimed to assess the chemical and thermal stability of the synthesized PP–COF, PT–COF, and PE–COF for potential practical applications. Our findings revealed that these COFs exhibited exceptional stability, rendering them highly desirable for applications requiring resistance to chemical reactions and elevated temperatures. Through thermogravimetric analysis (TGA), we observed that these COFs demonstrated outstanding thermal stability. Even at high temperatures, they maintained their structural integrity without significant weight loss or decomposition. Notably, these COFs exhibited no signs of breakdown until reaching 400 °C in a nitrogen environment. Furthermore, we conducted chemical stability tests on the PP–COF, PT–COF, and PE–COF samples in various solvents at room temperature over a 24-h period. These solvents included boiling water, ethanol,
N,N-dimethylformamide, dimethylsulfoxide, 3 M HCl at 25 °C, and 3 M NaOH at 25 °C. Surprisingly, post-testing, the PXRD patterns of the COF samples remained robust and unchanged, indicating the preservation of their remarkable crystallinity even under challenging conditions (
SI, Section S6, Figures S9–S11). This study confirms the robust chemical stability of PP–COF, PT–COF, and PE–COF, making them ideal candidates for functionalization. The appeal of these COFs as chemically and thermally stable materials is further amplified by their crystalline structure and porous attributes. To facilitate spectroscopic analysis and sensor uses, we grew thin films of PT–COF, PP–COF, and PE–COF on fused silica (
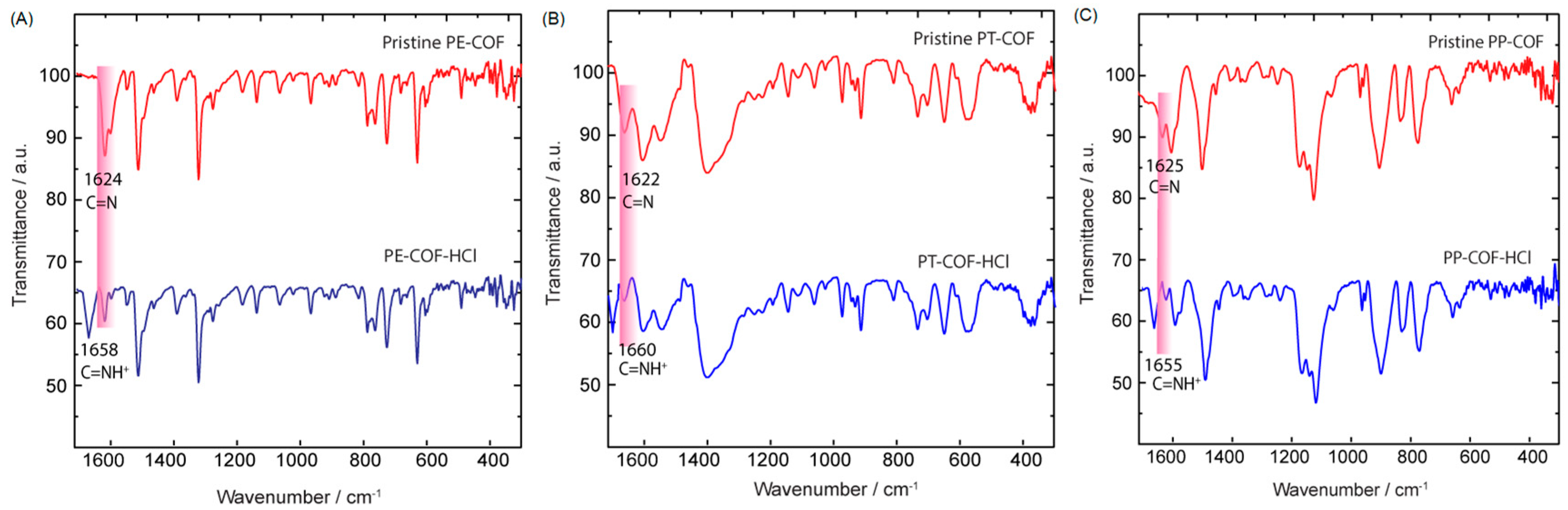
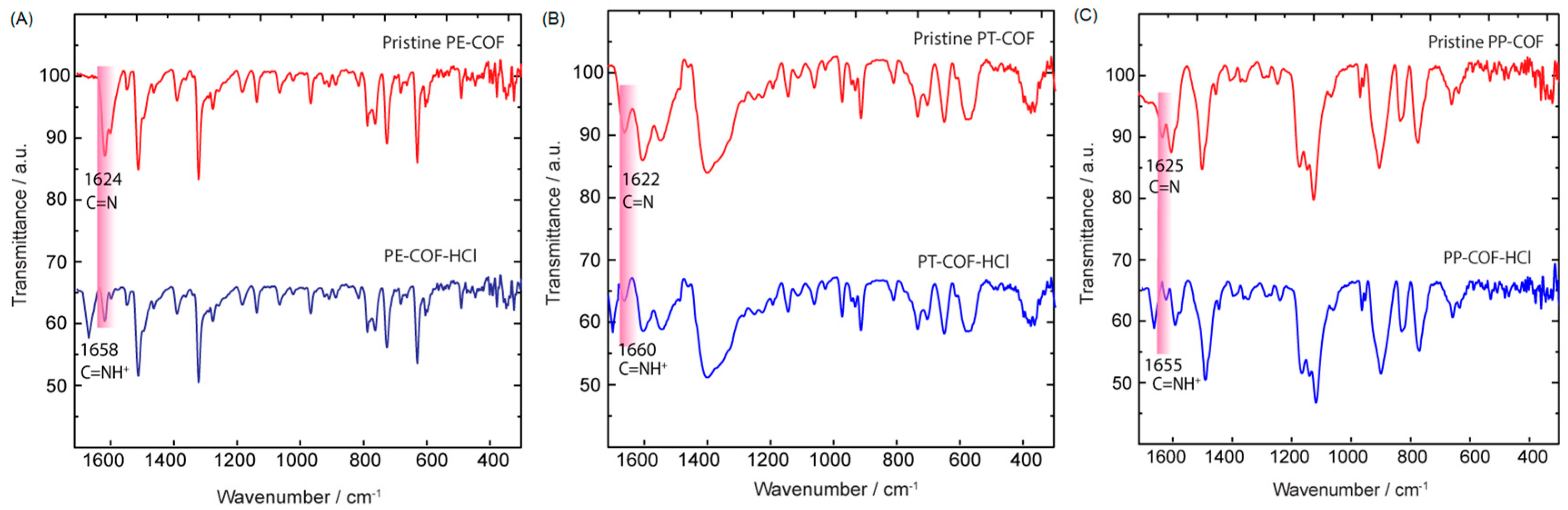
SI, Section S4). Despite the susceptibility of imine nitrogen atoms to strong acid protonation, the closely packed 2D COF structures provide stability against hydrolysis, enabling complete and reversible protonation even in humid conditions without notable degradation [
18]. To figure out the sensing mechanism of PT–COF, PP–COF, and PE–COF toward HCl, we recorded FT-IR spectrum of the sample of PT–COF, PP–COF, and PE–COF treated with gaseous HCl (termed PT–COF–HCl, PP–COF-HCl, and PE–COF-HCl). Compared with the IR spectra of pristine PE–COF, PT–COF, and PP–COF, the most apparent changes are highlighted in pink where new peaks at approximately 1658, 1660, and 1655 cm
−1 alongside a reduced intensity in the C=N band (1622–1625 cm
−1,
Figure 3A–C). These alterations signify the formation of protonated imine bonds (C=NH
+), a result of the swift protonation of imine nitrogen atoms by HCl gas [
19].
The photophysical characteristics of our COFs were investigated by analyzing their fluorescence emission spectra in a solution. The COFs were dispersed in 1,4-dioxane at a concentration of 1 mg mL−1, followed by excitation of the suspensions using light at a wavelength of 366 nm. The resulting fluorescence spectra of PT–COF, PE–COF, and PP–COF exhibited emission maxima at 450, 525, and 510 nm, respectively. These distinct emission peaks indicate the unique fluorescence behavior inherent in each COF variant. These features encouraged us to investigate the sensing properties of our COFs toward HCl.
Interestingly, upon exposure to HCl gas, suspensions of PT–COF and PP–COF (1 mg mL
−1) in 1,4-dioxane underwent a notable color shift, swiftly transitioning from yellow to dark brown. Subsequent exposure to triethylamine vapor facilitated the rapid restoration of the COF’s original yellow color, highlighting the remarkable reversibility inherent in the process of sensing gaseous HCl. Additionally, these color changes were visibly apparent and occurred within response times of less than 1 s—significantly faster than those of previously reported HCl sensors [
20]. A video showing the repeated protonation and deprotonation of a pyrene COF film (PP-COF) is supplied in the SI. Similar color transformations were observed in PE-COF from yellow to dark orange upon exposure to HCl vapor demonstrates strong interactions between our synthesized COFs and HCl molecules.
The reversibility of the color change remained uncompromised even after the COFs were subjected to 10 cycles of alternating treatments with HCl and triethylamine (TEA). This endurance underscores the robustness of the COFs in maintaining their reversible properties. Given the distinct and rapid color change that is perceptible without aid, this porous material stands as a commendable candidate for detecting HCl gas. To delve deeper into the structural impact of HCl gas followed by TEA gas exposure, the PT–COF, PP–COF, and PE–COF powders underwent analysis using powder X-ray diffraction (XRD) and Fourier-transform infrared spectroscopy (FT-IR). As depicted in (
SI, Section S7, Figures S12 and S13), minimal changes were observed in the major peaks of both the powder XRD patterns and IR spectra before and after exposure to HCl/TEA gas. These subtle alterations suggest that the fundamental framework of PT–COF, PP–COF, and PE–COF remained largely intact despite exposure to HCl/TEA gas, indicating their robustness in withstanding chemical exposure without significant structural damage.
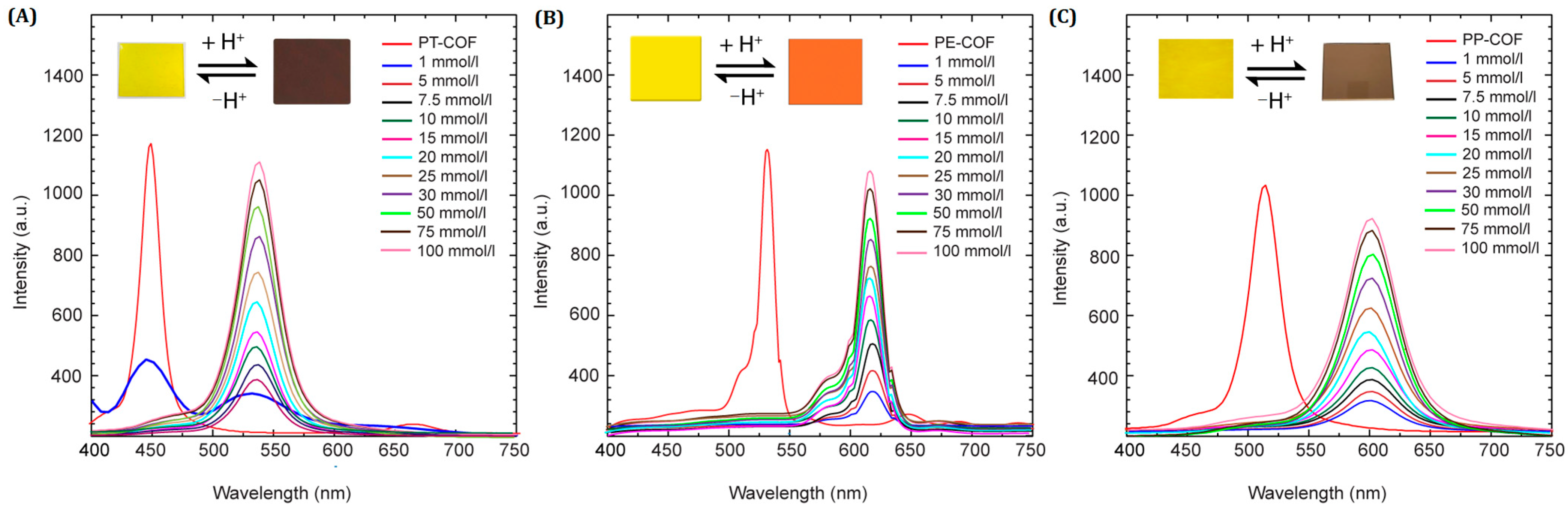
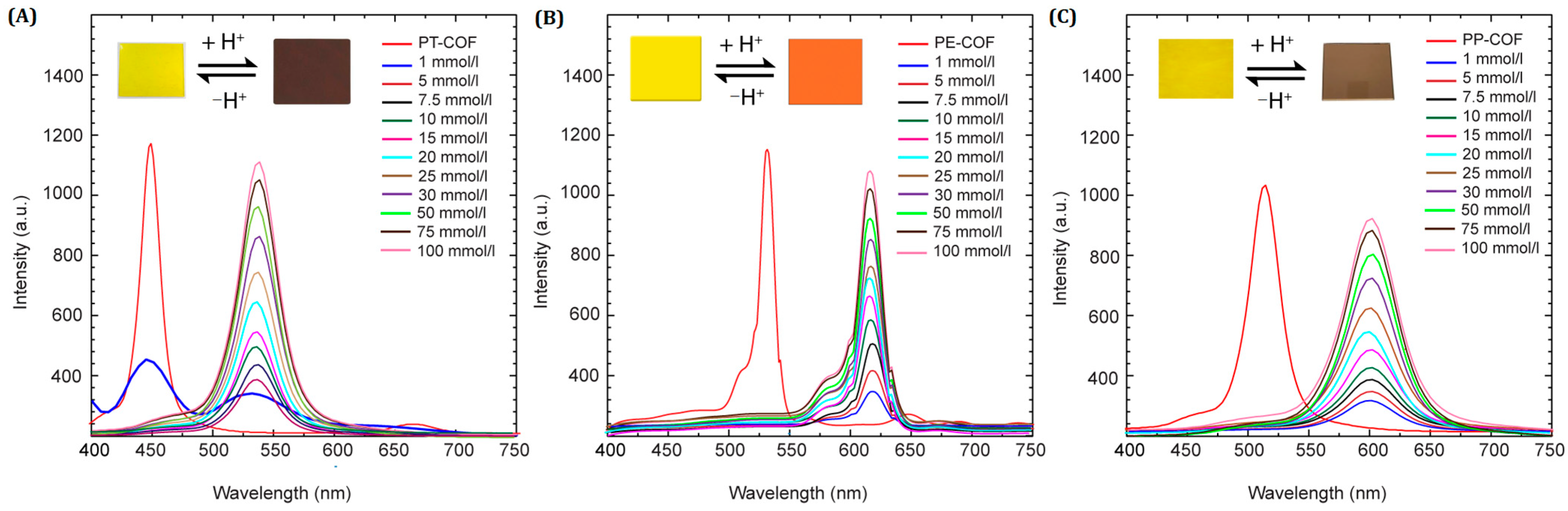
The influence of varying HCl concentrations on COF fluorescence was studied using a 1,4-dioxane solution of HCl due to challenges in precisely monitoring gaseous HCl concentrations. The fluorescence spectra of the PT–COF, PP–COF, and PE–COF suspensions in 1,4-dioxane revealed distinct responses to different HCl levels (
Figure 4). At 1 mmol L
−1 HCl, the fluorescence emission maxima significantly decreased at 450 nm, 525 nm, and 510 nm for PT–COF, PE–COF, and PP–COF, respectively, while new peaks emerged at 540 nm, 625 nm, and 611 nm. These signals vanished at 5 mmol L
−1 HCl, with subsequent gradual increases from 1 to 50 mmol L
−1, showing no further changes in emission. Calibration curves for HCl concentrations (1 to 50 mmol L
−1) exhibited linear correlations at 540 nm, 625 nm, and 611 nm, demonstrating detection limits of approximately 17, 10, and 20 nmol L
−1 for PT–COF, PE–COF, and PP–COF, respectively (
SI, Section S7, Figure S14).
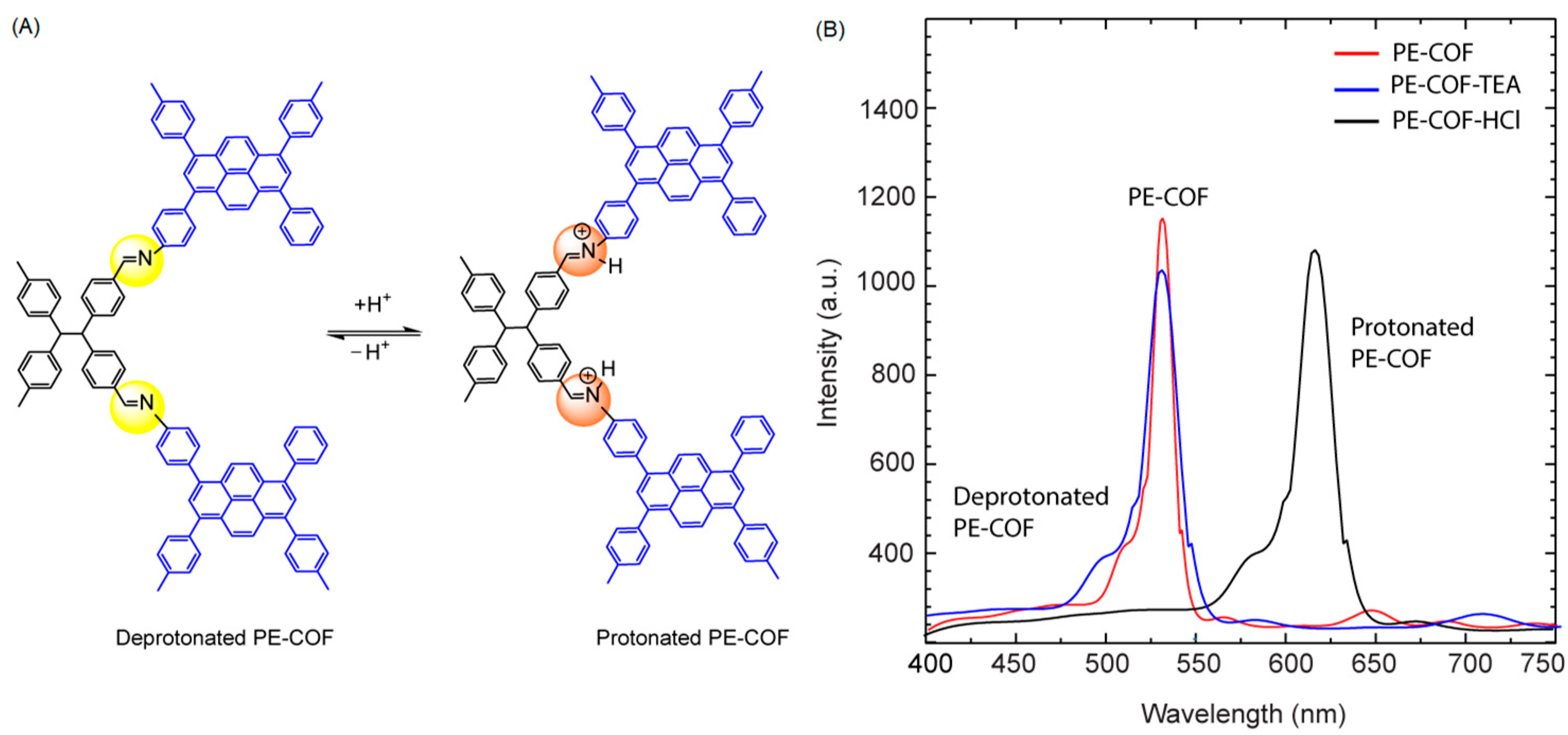
The sensing mechanism of PT–COF, PE–COF, and PP–COF toward HCl involves the probable protonation of imine nitrogen atoms within the COF skeletons when exposed to an HCl atmosphere. This protonation influences the conjugated structure of COF, leading to significant changes in fluorescence emission and color. Unlike many HCl-responsive materials found in the literature, where protonation sites typically locate at heteroaromatic nitrogen or amino units, in PT–COF, PP–COF, and PE–COF, protonation occurs on abundant and periodically distributed imine nitrogen atoms within the imine-linked COF skeletons. As demonstrated by Ascherl et al. [
15], the resulting protonated imine (C=NH
+) groups in COF structures act as stronger electron acceptors than their free imine (C=N) counterparts, accelerating charge transfer toward pyrene units, potentially causing a red-shift in transition photoexcitation energies. Specifically, PE–COF rapidly turned dark orange from yellow when exposed to HCl vapor (
Figure 5A), showcasing swift nitrogen atom protonation. Its fluorescence peak shifted from 525 nm to 625 nm with HCl and reverted to 525 nm post-TEA treatment (
Figure 5B).
Monitoring pH levels is crucial across various domains like chemistry, environment, and engineering. Two-dimensional COFs containing proton-donating/accepting units, exhibit pH-responsive behaviors driven by nitrogen atom protonation/deprotonation, altering color or fluorescence. These COFs, designed for sensing acid solution pH, also function as effective chemosensors for acidic gasses due to their porous structures that allow gaseous analyte diffusion. The pivotal protonation/deprotonation of imine bonds within COFs, influenced by agents like HCl and TEA, significantly affects their electronic properties, which is crucial for accurate acid sensing in acidic environments.
We delved into the impact of varying humidity levels on the detection capabilities of PT–COF, PP–COF, and PE–COF in the presence of HCl gas. Testing these materials across a spectrum of humidity environments (40%, 50%, 60%, 70%, and 80%) within a controlled constant temperature and humidity chamber (
SI, Section S8, Figure S15) revealed an intriguing outcome. Surprisingly, the fluorescence intensities of PT–COF, PP–COF, and PE–COF remained consistently unaffected by shifts in humidity levels while detecting HCl gas. Even under fluctuating humidity conditions, these materials consistently showcased steadfast and robust fluorescence responses. This resilience underscores the stability of their detection capacity, seemingly unswayed by changes in humidity. Further probing into the mechanisms behind this resilience might unveil prospects for deploying these materials in environments characterized by diverse humidity levels, expanding their range of potential applications.
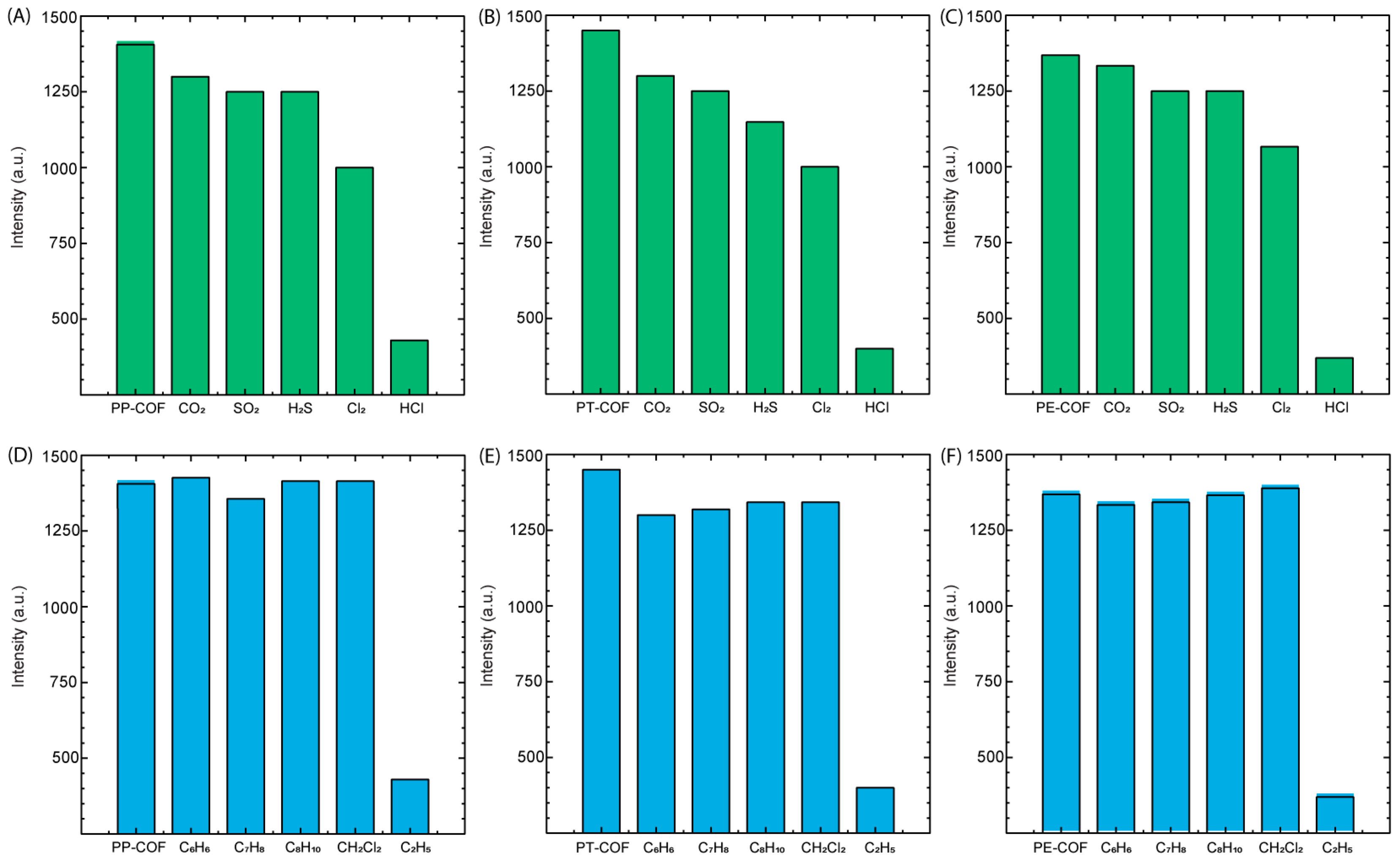
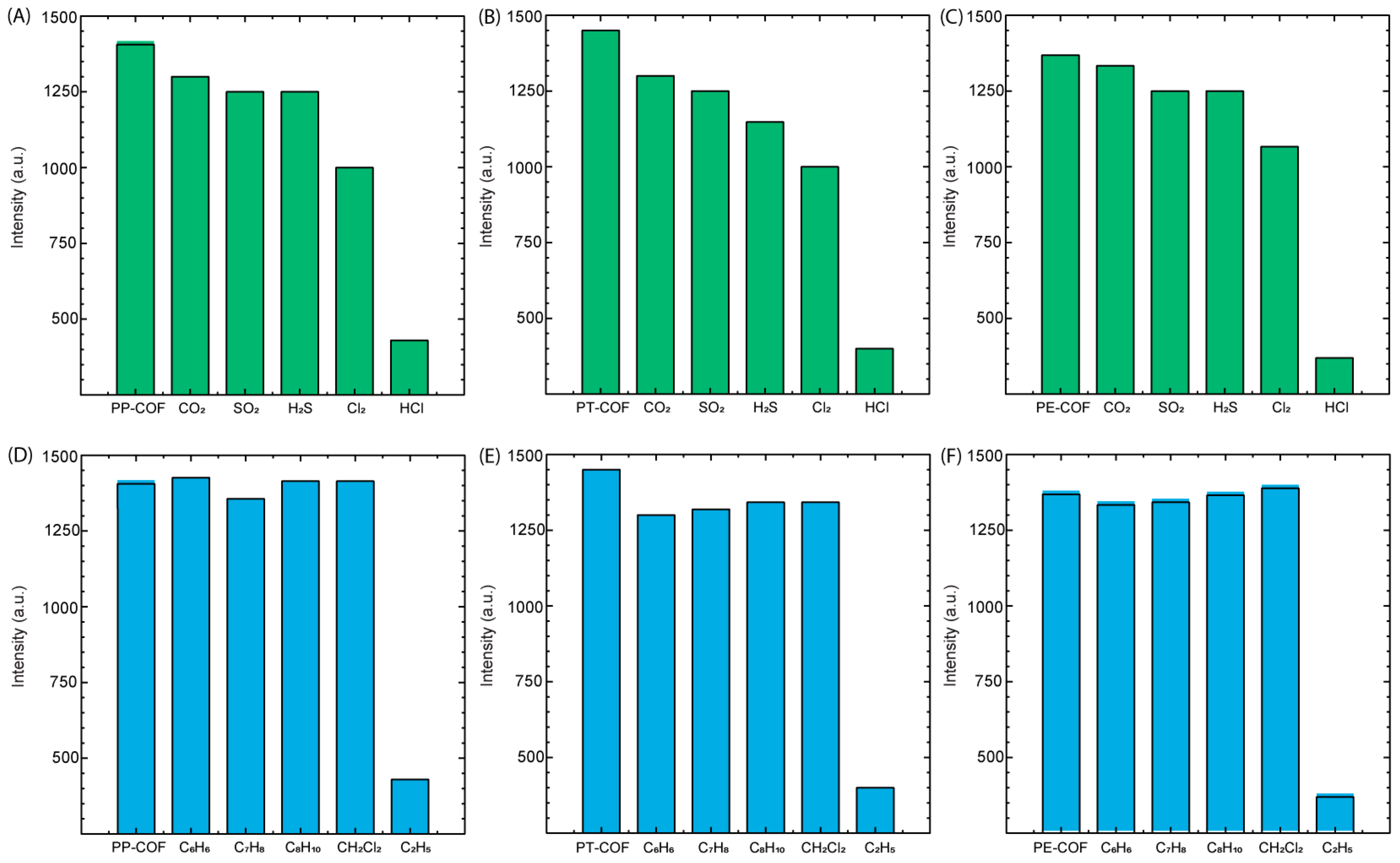
In the exploration of PT–COF, PP–COF, and PE–COF films’ reactivity towards co-existing acid gasses, our investigation was extended to evaluate their responses to CO
2, SO
2, H
2S, and Cl
2. As depicted in
Figure 6A–C, exposure to CO
2 and SO
2 resulted in no discernible changes in fluorescence intensity within the COF films. However, the presence of H
2S and Cl
2 induced a modest reduction in fluorescence intensity, by approximately ~20%. The discrepancy in fluorescence reduction induced by H
2S and Cl
2 compared to gasses like CO
2 and SO
2 may be attributed to their distinct chemical reactivity and interactions with the COF materials. H
2S and Cl
2 exhibit heightened chemical reactivity, with H
2S being renowned for its reducing properties and Cl
2 being a potent oxidizing agent. These characteristics likely prompt more substantial chemical interactions with the COF, potentially altering fluorescence intensity. Additionally, H
2S and Cl
2 might possess specific binding affinities or interactions with functional groups within the COF structure, potentially influencing electronic states or modifying emission properties, thereby accentuating the observed decrease in fluorescence intensity. Notably, these findings underscore the COFs’ limited reactivity towards certain gasses while indicating a moderate interaction with hydrogen sulfide and chlorine gas. Importantly, amidst these varied responses, the COF materials exhibited a distinctive and consistent reactivity to hydrogen chloride (HCl), demonstrating selectivity in detecting this specific acid gas. This observed selectivity emphasizes the potential utility of PT–COF, PP–COF, and PE–COF for targeted and precise detection of hydrogen chloride, elucidating their applicability in scenarios where specific gas identification is paramount.
In our investigation, special attention was given to the role of volatile organic compounds (VOCs) as potential interfering agents in the detection of hazardous gasses. The influence of four commonly encountered VOCs—benzene (C
6H
6), toluene (C
7H
8), xylene (C
8H
10), and dichloromethane (CH
2Cl
2)—was meticulously evaluated for their impact on the fluorescence-based detection capabilities of three covalent organic frameworks (COFs): PT–COF, PP–COF, and PE–COF. The experimental outcomes, as depicted in
Figure 6D–F, reveal a remarkable stability in the fluorescence response of the COFs upon exposure to these VOCs. Notably, fluorescence intensity remained largely unchanged, indicating a negligible interaction between the COFs and the VOCs. This observation is particularly significant, as it suggests that the molecular architecture of PT–COF, PP–COF, and PE–COF provides a selective pathway for hydrogen chloride gas detection, effectively mitigating the potential cross-sensitivity issues commonly associated with the presence of VOCs in environmental and industrial settings. These findings underscore the potential of COFs in advancing gas sensing technology. Their ability to selectively detect target gasses amidst interfering VOCs without losing sensitivity or specificity is crucial for environmental monitoring, industrial safety, and public health. The stability and selectivity demonstrated by PT–COF, PP–COF, and PE–COF pave the way for further research to broaden the application of COF-based sensors in diverse detection environments.
In our study, we assessed the selectivity of PT–COF, PP–COF, and PE–COF for hydrogen chloride (HCl) by exposing their suspensions to different acids at a concentration of 20 mmol L
−1. The results, detailed in the
Supplementary Materials (SI), Section S9, Figure S16, revealed that only HCl triggered a significant redshift in the fluorescence emission peaks of these COFs to 540 nm for PT–COF, 625 nm for PP–COF, and 611 nm for PE–COF, along with a substantial increase in fluorescence intensity. This indicates that these COFs are highly selective for HCl, showcasing a distinct fluorescence response hierarchy: HCl > HBr > H
2SO4 > HNO
3 > H
3PO
4 > CH
3COOH.
This specificity sequence suggests that the response of COFs to various acids is influenced by the strength of the acids and their interaction with COFs. It appears that the ability of an acid to protonate the functional groups within the COF structure plays a critical role. This protonation alters the COFs’ electronic structure, affecting their fluorescence properties. Strong acids like HCl, which are proficient proton donors, induce notable changes in the electronic configuration of the COFs, resulting in more significant fluorescence shifts. In contrast, weaker acids such as acetic acid (CH3COOH) have less impact, likely due to their lower proton-donating capacity, leading to minimal changes in the COFs’ structure and fluorescence response. This behavior underscores the potential of these COFs as selective sensors for detecting HCl, offering insights into designing advanced materials for gas sensing applications.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}