Abstract
Macrolides are widely used in medicine and veterinary medicine, and are the leading antibiotics in terms of consumption. The release of macrolides and their metabolites into the environment through municipal wastewater can have an adverse effect on aquatic ecosystems and human health. In the present study, a method for the non-targeted screening and semi-quantitative determination of macrolide antibiotics and their derivatives in wastewater based on a combination of chromatographic separation and tandem mass spectrometric detection in precursor ion scan (PrecIS) mode has been proposed. Product ions with m/z 158 and 174 related to specific desosamine fragments were used as diagnostic ions for the PrecIS detection of the macrolide structures without (14- and 15-membered macrocycles) and with a (16-membered macrocycle) glycosylated desosamine moiety, respectively. The combination of the optimized solid phase extraction procedure and HPLC-MS/MS analysis in PrecIS mode allowed for the suspect screening of macrolides in municipal wastewater with limits of detection in the range of 4–150 ng L−1. The developed approach made it possible to detect and tentatively identify in municipal wastewater 17 compounds belonging to the macrolide class, including azithromycin, clarithromycin, josamycin and 14 metabolites with a total concentration of 1450 ng L−1.
1. Introduction
Macrolide antibiotics (or macrolides) are a large group of compounds consisting of a 14-, 15- or 16-membered macrocyclic lactone ring to which one or more carbohydrate moieties (including desosamine, playing a key role in antibacterial action) are attached. Macrolides possess a broad spectrum of activity against many gram-positive bacteria and are widely used in medicine and veterinary medicine [1,2], being the leaders in terms of consumption among all antibiotics [3,4,5]. The consumption of macrolides has especially increased during the COVID-19 pandemic due to concerns about bacterial co-infection [6,7,8,9]. The variety of synthesized and used in practice macrolides is also rapidly growing due to the need to avoid the emergence of bacterial resistance to antibiotics. For example, in recent work by Seiple et al. [10], more than 300 structurally diverse macrolide antibiotic candidates were prepared, and the majority of these structures had antibiotic activity.
Antibiotics are not completely metabolized after therapeutic use and are excreted (usually 55–80%) from the body as a mixture of parent compounds and their active metabolites [11]. Since conventional wastewater treatment processes are not able to effectively remove such pollutants [12], macrolides enter the environment through municipal wastewater adversely affecting aquatic ecosystems. Moreover, it has been shown that wastewater treatment plants can serve as potential reservoirs of antibiotic resistance genes that can be transmitted to human-associated bacteria through water and food chains [13,14]. As a result, studies on the occurrence, levels, fate and effects of antibiotics (including macrolides) in aquatic environments have increased in recent years [15].
Many studies and several comprehensive reviews [16,17,18] devoted to solving the urgent problem of assessing macrolides in wastewater and natural reservoirs are available in the literature. According to them, the sample preparation typically involves the solid-phase extraction (SPE) of macrolides on an HLB polymeric functionalized sorbent (co-polymer of divinylbenzene and N-vinylpyrrolidone) with high sorption capacity towards both non-polar and moderately polar analytes providing recoveries in the range of 60–90% [19,20,21]. The quantification of macrolides in extracts with complex matrices was carried out by high performance liquid chromatography–electrospray ionization (ESI) tandem mass spectrometry (HPLC-MS/MS) [22,23,24] as the most selective analytical technique. For the chromatographic separation of macrolides, reverse-phase stationary phases (C18 and C8) were successfully used in combination with mobile phases consisting of mixtures of water with acetonitrile or methanol [25,26].
It is worth noting that existing analytical approaches involve the targeted determination of analytes in a multiple reaction monitoring (MRM) mode and require corresponding analytical standards. At the same time, unexpected macrolide antibiotics or their metabolites can be present in municipal wastewater, while corresponding analytical standards can be commercially unavailable or hardly available. To overcome this problem, non-targeted analytical methods for macrolide screening and standard-free determination must be developed. Due to the fact that macrolides have similarities in their structures, they can produce the same specific product ions during collision-induced dissociation (CID) in tandem mass spectrometry analysis. For this reason, the use of an HPLC-MS/MS technique in a precursor ion scan (PrecIS) providing both non-target and target screening capabilities is considered most promising. This technique is typically implemented on triple quadrupole mass analyzers and involves the detection of a specific product ion at a fixed m/z on a third quadrupole (Q3) while scanning a wide m/z range of precursor ions on a first quadrupole (Q1). According to the literature, this approach has been successfully applied in the non-targeted analyses of various complex objects: for screening estradiol esters in cattle hair [27], pentacyclic triterpenoids in plants [28] and benzimidazoles in the serum [29], etc.
Thus, this work is aimed at developing an approach to the screening and semi-quantitative determination of macrolides of various classes by high performance liquid chromatography-PrecIS tandem mass spectrometry and its application to the study of real samples of municipal wastewater.
2. Materials and Methods
2.1. Reagents and Materials
Six commercially available 14-, 15- and 16-membered macrolides were used in the study (Figure 1): azithromycin (>98%) and erythromycin (>98%) purchased from Sigma-Aldrich (Steinheim, Germany); clarithromycin (>96%), spiramycin (>90%) and josamycin (>98%), purchased from Alfa Aesar (Heysham, UK); and midecamycin (>85%) purchased from Toronto Research Chemicals (North York, ON, Canada).
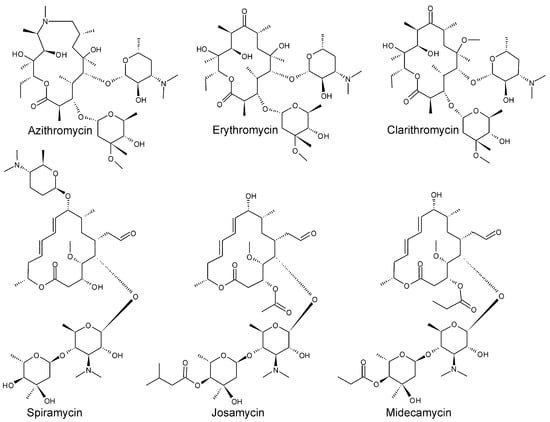
Figure 1.
Structural formulas of the studied macrolides.
High Performance Liquid Chromatography-gradient grade methanol (Merck, Darmstadt, Germany), “type I” Milli-Q high-purity water, formic acid (≥96%) and ammonium formate (10 M aqueous solution) purchased from Sigma-Aldrich (St. Louis, MO, USA) were used in sample preparation procedures and as components of the mobile phase in HPLC-MS analyses.
Stock solutions of analytes in methanol with concentrations of 250 mg L−1 were prepared from precisely weighed portions. The calibration solutions with concentrations of 10–10,000 μg L−1 were obtained by mixing and sequentially diluting the stock solutions with 20% aqueous methanol.
2.2. Municipal Wastewater Sample Preparation
Urban wastewater was collected in March 2021 at the municipal wastewater treatment plant in Arkhangelsk (Russia) from the point of discharge into the natural reservoir (Khatoritsa River). The sample was taken in a 2.5 L amber glass bottle, then immediately delivered to the laboratory and subjected to solid phase extraction (SPE) according to the EPA 1694 method [30] with some modifications. Since this method was designed for the simultaneous analysis of the widest possible range of pharmaceuticals and personal care products, it involves extraction at two pH levels (2 and 12) not optimal for macrolides. For this reason, in the present study the pH was adjusted to 4, which ensured the most efficient extraction of analytes [11,31]. In addition, the use of isopropanol as an elution solvent instead of methanol allowed for a noticeable increase in elution completeness and thus achieving the satisfactory analyte recoveries of 53–94% (Supplementary Material, Table S1). Expectedly, the lowest recoveries (<60%) were observed for the most polar analytes possessing a tertiary amine group in a lactone ring (azithromycin) or an additional desosamine moiety (spiramycin). The recoveries of the other tested compounds exceeded 73%.
Two hundred milligrams of Na4EDTA × 2H2O was added to 500 mL of pre-filtered wastewater brought to pH 4. An SPE was carried out in a Chromabond HLB (Macherey-Nagel, Duren, Germany) 3 mL cartridge filled with 200 mg of hydrophilic-lipophilic balanced N-vinylpyrrolidone-divinylbenzene sorbent on an AH0-6023 vacuum manifold (Phenomenex, Torrance, CA, USA). Before extraction, an SPE cartridge was conditioned with 20 mL of isopropanol and 6 mL of deionized water at pH 4. A wastewater sample was passed at a flow rate of 5–10 mL min−1, then the sorbent was flushed with 10 mL of deionized water and air-dried for 15 min with vacuum pump running. The analytes were eluted with 12 mL of isopropanol. The obtained extract was evaporated in a conical glass vial at 50 °C to a volume of 100 μL under a nitrogen stream, then 400 μL of deionized water was added. Thus, the reached pre-concentration degree was 1000.
2.3. Liquid Chromatography–Tandem Mass Spectrometry
An LCMS-8040 HPLC-MS/MS system (Shimadzu, Kyoto, Japan) consisting of an LC-30 “Nexera” liquid chromatograph and triple quadrupole mass analyzer was used in liquid chromatography–tandem mass spectrometry analyses. The HPLC system consisted of a DGU-A5 vacuum degasser, two LC-30AD chromatographic pumps, an LC-30AC autosampler and a CTO-30A column thermostat. The system control and data analysis were performed using LabSolutions 5.56 software (Shimadzu, Kyoto, Japan).
Chromatographic separation was achieved in a gradient elution mode on a Nucleodur PolarTec column (Macherey-Nagel, Duren, Germany), 150 × 2.0 mm, particle size 1.8 μm, with a reverse-phase sorbent with embedded amide groups providing additional selectivity for polar compounds. The sample injection volume was 2 μL, mobile phase flow rate 0.3 mL min−1 and column thermostat temperature 40 °C. The mobile phase consisted of solvent A (deionized water with 0.1% formic acid and 5 mM ammonium formate) and solvent B (acetonitrile with 0.1% formic acid). The gradient elution was programmed as follows: 0–2 min—10% B, 2–10 min—linear ramp to 50% B, 10–15 min—50% B.
Mass spectrometry detection was carried out in MRM and PrecIS modes using electrospray ionization in a positive ion mode (ESI+). Ion source parameters: spraying, drying and curtain gas (nitrogen) flow rates—2 and 7.5 L min−1, respectively; desolvation line and heat block temperature—300 °C and 400 °C, respectively; ESI capillary voltage—4.5 kV. A collision-induced dissociation (CID) with argon as a collision gas was used for obtaining tandem mass spectra. The conditions for mass spectrometric detection of macrolides in MRM mode were optimized in an automatic regime by the flow injection of analyte standard solutions (Table 1).

Table 1.
Optimized conditions for mass spectrometric detection of six macrolide antibiotics in the MRM mode.
In the PrecIS mode, the precursor ion scanning range was m/z 400–1200, with the scan frequency of 10 Hz.
2.4. Liquid Chromatography–High-Resolution Mass Spectrometry
Liquid chromatography–high-resolution mass spectrometry (HPLC-HRMS) analyses were carried out using an HPLC-HRMS system consisting of the same HPLC system as described in Section 2.3 and an Orbitrap ID-X high-resolution “tribrid” mass spectrometer (Thermo Scientific, Waltham, MA, USA) equipped with an OptaMax NG ion source with ESI probe. Chromatographic separation was carried out within the same parameters as described in Section 2.3. In mass spectrometry detection, the following parameters were applied: spraying, drying and curtain gas flow rates—50, 10 and 3 arb. units, respectively; transfer line temperature—325 °C; ESI capillary voltage—4.5 kV. The spectra were scanned in an m/z range of 200–1200 using an orbital ion trap mass analyzer with resolving power set to 120,000 (M/ΔM, at m/z 200). To maintain the highest accuracy in determining m/z (<3 ppm), internal EASY-IC mass scale calibration was used. Xcalibur software (Thermo Scientific, Waltham, MA, USA) was used to control the instrument and acquire HRMS chromatograms.
In the study of the analytes’ mass spectrometry fragmentation and in attributing the peaks in the obtained tandem mass spectra (Section 3.1), the flow injection of analyte solutions was used with the further determination of the elemental compositions of the product ions based on their accurate masses. A higher energy collisional dissociation (HCD) similar to CID in triple quadrupole instruments was used. The fragmentation pathways were proposed using ACD Fragmenter software (ACD/Labs, Toronto, ON, Canada).
2.5. Quantification and Method Validation
The quantification of the target analytes in both MRM and PrecIS detection modes was carried out by an external standard method with calibration curves constructed using the calibration solutions of the six macrolides (Section 2.1). The same approach was applied to the semi-quantitative assessment of the detected macrolides or their metabolites by HPLC-MS/MS with PrecIS detection using the calibration curves obtained for the standards structurally close to the corresponding analytes. Limits of detection (LOD) and quantification (LOQ) were determined using the signal-to-noise ratio criteria of 3 and 10, respectively, and then refined in the analysis of the solution with analyte concentration close to LOQ. The full method validation, involving an estimation of matrix effects, intra-day and inter-day accuracy and precision, cannot be considered applicable in the case of the screening studies in the absence of the corresponding analytical standards.
3. Results and Discussion
3.1. Tandem Mass Spectra of Macrolides and Selection of Diagnostic Product Ions
In developing a method for the non-targeted screening of macrolides in PrecIS mode, a key role is played by the choice of a diagnostic ion in tandem mass spectra that is specific for a given class of compounds and is characterized by a high signal intensity to achieve an acceptable sensitivity of the analysis. During CID, the protonated molecules ([M + H]+) of six analytes with m/z values listed in Table 1 give a number of product ions (Figure 2) associated with the elimination of sugar and desosamine moieties and the cleavage of the macrocyclic lactone ring (Supplementary Material, Figures S1–S6). The latter proceeds quite easily even at low collision energies, resulting in the absence of noticeable signals of characteristic macrocyclic fragments of macrolides in MS/MS spectra. In this situation, only the desosamine fragment can be considered as a suitable diagnostic ion, which gives intense signals in the mass spectrum due to the presence of an easily protonated dimethylamine group. It is worth noting that the CID of the macrolides containing an unsubstituted desosamine moiety in their structure (azithromycin, erythromycin and clarithromycin) leads to the formation mainly of the desosamine-related product ion (dehydrated desosamine molecule) with m/z 158.1175 and an elemental composition [C8H15NO2 + H]+, while the other three 16-membered macrocyclic compounds with carbohydrate substituents attached to the desosamine ring give an intense signal at m/z 174.1125 attributed to the protonated molecule of 4-(dimethylamino)-2-methyl-3,4-dihydro-2H-pyran-3,5-diol or dehydrated molecule of micaminose with an elemental composition [C8H15NO3 + H]+ (Figure 2). The latter is formed as a result of the cleavage of the ether bond between the desosamine ring and sugar moiety (Supplementary Material Figures S4–S6).
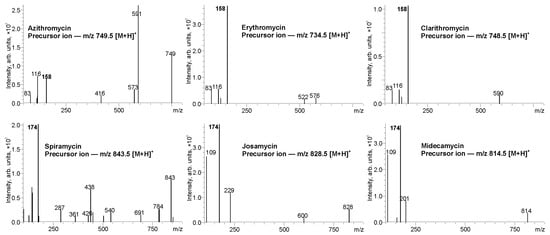
Figure 2.
Tandem mass spectra of macrolides (collision energy 30 eV).
Thus, the detection of the different characteristic product ions for the two types of macrolide structures opens possibilities for their mass spectrometric discrimination and non-targeted screening by PrecIS tandem mass spectrometry. The fragments with m/z 158 and 174 can be used as diagnostic product ions for the selective group detection of the structures without (14- and 15-membered macrocycles) and with a (16-membered macrocycle) glycosylated desosamine moiety. To ensure a highest sensitivity of the analysis, CID collision energy has been optimized by varying in the range of 5–60 eV using the triple quadrupole mass analyzer. The highest signal intensities of the selected diagnostic ions with m/z 158 and 174 were achieved at the values of 32 and 36 eV, respectively, which were used for further method development.
3.2. Method Development and Validation
Due to the polar nature of the analytes, their chromatographic separation on common octadecyl silica reverse-phase HPLC columns is challenging. During preliminary experiments, it was found that acceptable separation selectivity can be attained using a Nucleodur PolarTec stationary phase with embedded polar amide groups that increase retention and separation selectivity due to the combination of hydrophobic and polar interactions with the analytes. The best results (Figure 3) were obtained in a gradient elution mode with the mobile phase consisting of water and acetonitrile with the addition of 0.1% HCOOH and 5 mM ammonium formate. The addition of ammonium salt allowed the suppression of undesirable interactions of the analytes with residual silanol groups of the sorbent, leading to the distortion and broadening of the chromatographic peaks of azithromycin and spiramycin.
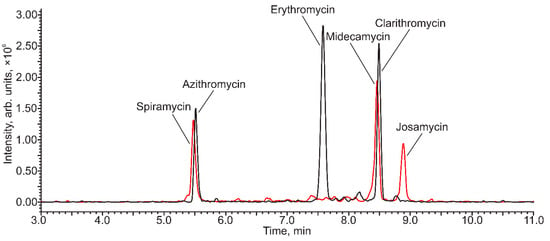
Figure 3.
The HPLC-MS/MS (PrecIS) chromatogram of the model mixture of macrolide antibiotics (Azithromycin and spiramycin—500 µg L−1, erythromycin, midecamycin, clarithromycin and josamycin—50 µg L−1) obtained with the detection of diagnostic product ions with m/z 174 (red) and m/z 158 (black).
As can be seen from Figure 3, although two pairs of analytes (azithromycin-spiramycin and clarithromycin-midecamycin) were not fully separated by HPLC, their selective detection was not problematic due to the difference in diagnostic product ions. Moreover, noticeable interference in the quantification of these analytes in their simultaneous presence was not observed.
Being a scanning detection mode, PrecIS is significantly inferior in sensitivity to MRM. However, the attained instrumental LODs and LOQs for most of the analytes presented in Table 2 turned out to be rather low (~10 µg L−1).
Table 2.
Parameters of the calibration curves (y = sx + a) for the dependence of the chromatographic peak area (y) on the concentration of analyte (x); instrumental and method LODs and LOQs for the determination of six macrolides by HPLC-MS/MS in the PrecIS mode.
This can be explained by the high ionization efficiency of the nitrogen-containing compounds and lower noise level in the monitored precursor ion m/z range (400–1200). The exceptions are azithromycin and spiramycin, both possessing two nitrogen atoms in their structures. Their instrumental LODs were one order of magnitude higher than those of other analytes, which is probably due to the formation of doubly charged ions or partial degradation in the ion source. Nevertheless, in the combination with an appropriate sample preparation (pre-concentration) procedure, the sensitivity of the PrecIS detection can be considered sufficient to ensure the reliable detection of the analytes in real samples. The pre-concentration degree (up to 1000×) and recoveries provided by SPE (Section 2.2) on an HLB polymeric sorbent allowed for the detection of the analytes in water samples with method LOQs in the range of 13–21 ng L−1 (400 and 490 ng L−1 for azithromycin and spiramycin, respectively) with the linear range covering at least two orders of magnitude (Table 2). These values were additionally confirmed in the analyses of model water samples with analyte concentrations close to LOQ (Supplementary Material, Figure S7).
3.3. Targeted Analysis of Municipal Wastewater
To evaluate the accuracy of macrolide PrecIS quantification in real municipal wastewater, the concentrations of the six target analytes were determined in the studied sample by HPLC-MS/MS using both PrecIS and MRM modes. The comparison of the obtained results (Table 3) showed that the difference in the concentrations measured by the two methods did not exceed 20%, although RSD for clarithromycin and josamycin were up to one order of magnitude higher in PrecIS detection mode since these compounds were detected at the levels (21 and 14 ng L−1, respectively) close to LOQ. In total, out of the six target analytes, only three compounds were detected in the wastewater sample by PrecIS. In addition to the above-mentioned minor compounds, this number included azithromycin, which was the dominant component with a concentration two orders of magnitude higher compared to clarithromycin and josamycin. The higher sensitivity provided by the MRM mode allowed for the detection of the two additional macrolides present at the ng L−1 level: erythromycin and midecamycin. Spiramycin was not found in the studied wastewater by either the PrecIS or MRM detection mode.
Table 3.
Levels of six target macrolides in municipal wastewater determined by HPLC-MS/MS in PrecIS and MRM detection modes.
3.4. PrecIS Suspect Screening of Macrolides in Municipal Wastewater
The main advantage of the PrecIS tandem mass spectrometry mode over MRM is the possibility of the non-targeted detection of structurally close compounds giving specific diagnostic product ions in CID. The obtained HPLC-MS/MS chromatogram of the studied municipal wastewater extract (Figure 4) demonstrated the presence of a number of peaks with retention times in the range of 5–11 min.
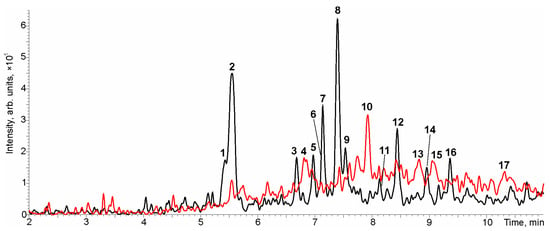
Figure 4.
The HPLC-MS/MS chromatogram of urban wastewater obtained in PrecIS mode using a product ion with m/z 174 (red) and a product ion with m/z 158 (black).
The manual search of precursor ions for each peak and obtaining their tandem mass spectra in additional chromatographic runs allowed the identification of 17 potential candidates (Table 4) including 12 compounds giving the diagnostic product ion with m/z 158 and only 5 compounds detected at m/z 174. Naturally, among them there were also three compounds (Nos. 2, 12 and 13) identified in the course of targeted analysis using standards. The chromatographic peaks not included in Table 4 had low (at the noise level) signal intensity or precursor ions with m/z values that did not relate to macrolides.
Table 4.
Tentative identification of macrolides in municipal wastewater by HPLC-MS/MS in PrecIS mode and HPLC-HRMS.
The obtained tandem mass spectra (Supplementary Material, Table S2) and available literature data [32,33,34,35,36,37,38] allowed for the tentative identification of the detected compounds. Their correctness was confirmed by an additional study by HPLC-HRMS (Supplementary Material, Figure S8) with the establishment of the exact m/z values and elemental compositions of the corresponding protonated molecules (Table 4).
Compound Nos. 6, 7, 11 and 14, giving a specific erythromycin product ion with m/z 558 in tandem mass spectra, were identified as erythromycin derivatives. Compound No. 7, with the molecular formula C38H71O14N, was assumed to be 10-hydroxy-4′-(2-hydroxyethylmethylamino)-erythromycin according to the Chemspider database. Compound No. 11 (C37H65O12N) can be considered as a product of the water molecule elimination from erythromycin (C37H67O13N) and thus can be attributed to anhydroerythromycin. The latter is known as a major product of erythromycin degradation in the acidic environment (stomach) and does not possess antibacterial activity [32]. The third representative of this group, compound No. 6, had an m/z identical to clarithromycin. However, since the specific clarithromycin fragment with m/z 590 was not observed in the tandem mass spectrum, it can be considered as an erythromycin derivative with the methylated hydroxy group in a carbohydrate moiety. The identification of compound No. 14 was the most difficult. Having the same elemental composition and a number of the same characteristic peaks in the tandem mass spectrum as erythromycin, it has a markedly higher chromatographic retention. This compound cannot be considered a product of the demethylation of the desosamine or cladinosyl moieties of clarithromycin, since the signals of desosamine (m/z 158) and the lactone ring without a methoxy group (m/z 576) were observed in the tandem mass spectrum. In this regard, compound No. 14 was assigned to the known [33] erythromycin transformation product, pseudo-erythromycin A-6,9-hemiketal (CAS 105900-46-7).
Compound Nos. 1, 3, 5, 8, 9 and 16 were classified as clarithromycin derivatives. Metabolite No. 3 (C30H55O10N) has the same elemental composition as a major clarithromycin fragment ion with m/z 590 (Supplementary Material, Figures S1 and S3) and appears to be a product of the hydrolytic elimination of the carbohydrate moiety (cladinosyl) from the parent macrolide. Earlier, this compound was described in the literature as an inactive clarithromycin degradation product [34]. The tandem mass spectrum of compound No. 8 contained an intense signal at m/z 606 differing from the above-mentioned clarithromycin fragment with m/z 590 by an additional hydroxy group. It can be assumed that the latter is attached to a carbon atom in the 14th position of a lactone ring [35], forming a metabolite with pronounced antibacterial activity [34]. Compound No. 9 has the same elemental composition as No. 8 (C38H69O14N) and a significantly different tandem mass spectrum. Its exact structure could not be established, but it can be assumed that the difference from No. 8 lies in the position of the hydroxy group. According to an elemental composition (C30H55O11N), compound No. 1 was identified as 3-O-decladinosyl-14-hydroxyclarithromycin, which was formed during the hydrolysis of 14-hydroxyclarithromycin by analogy with the formation of metabolite No. 3 from clarithromycin. Due to low signal intensity, an informative tandem mass spectrum of compound No. 16 (C35H61O11N) has not been registered. According to the Chemspider database, this compound was tentatively identified as a 12-allyloxy-3-O-decladinosyl-calrithromycin derivative (ChemSpider ID 9620153) with an acetylated desosamine moiety. The only alternative offered by the database is also a 14-membered macrolide (ChemSpider ID 28282589) with attached desosamine and a slightly modified carbohydrate moiety the origin of which is difficult to explain. A similar situation with the impossibility of obtaining a tandem mass spectrum was observed for compound No. 5 (C38H71O15N). According to Gago-Ferrero et al. [36], this compound is one of the clarithromycin metabolites. It is formed from clarithromycin through the cleavage of the C-O bond in the lactone ring (PubChem CID 139596847).
It is assumed that the remaining four compounds (Nos. 4, 10, 15 and 17) are derivatives of josamycin, since their tandem mass spectra (except for No. 4) contained the 4-O-isovalerylmycarose fragment with m/z 229 characteristic of this macrolide antibiotic. Differing from josamycin only in the number of oxygen atoms in the molecule, compound Nos. 10, 4 and 15 were identified as mono-, di- and tetrahydroxy derivatives, respectively. This is in a good agreement with the fact that the main metabolic pathway of josamycin is hydroxylation, resulting in the introduction of hydroxy groups both in the lactone ring and 4-O-isovalerylmycarose moiety [37,38]. The signals of the product ions formed as a result of the cleavage of 4-O-isovalerylmycarose in the tandem mass spectra showed that compound Nos. 10 and 15 possess the hydroxy groups only in the lactone ring, while compound No. 4 is distinguished by the presence of one hydroxy group in the lactone ring and the other is attached to the carbohydrate moiety. An exact structure of the 16-membered josamycin derivative No. 17 (C45H73O20N) with the highest molecular weight remained unclear and could be established only in higher order tandem mass spectrometry experiments, which were beyond the scope of the study.
3.5. Semi-Quantfication and Levels of Macrolides in Municipal Wastewater
Since the responses of target analytes turned out to be very different while structurally close compounds showed comparable ionization efficiency (Section 3.2), the standard-free semi-quantification of the detected metabolites requires the establishment of response factors for each class of macrolides. To overcome this problem, in the present study the calibration dependencies and extraction recoveries obtained for the parent macrolides (erythromycin, clarithromycin and josamycin) were used for the quantification of the corresponding metabolites. The obtained results (Table 4) demonstrate that the concentrations of the target compounds and detected metabolites lie in a relatively narrow range of 4–50 ng L−1, covering one order of magnitude. An exception is azithromycin, dominating among all the macrolides and accounting for more than 80% of their total content in the municipal wastewater (1450 ng L−1). The detected level exceeded the predicted no-effect concentration given in the literature [39]. This allowed the conclusion that at the site of the wastewater discharge in Arkhangelsk city there is a risk of the development of antibiotic resistance genes.
4. Conclusions
The tandem mass spectra of macrolide antibiotics are distinguished by intense signals of the desosamine fragments with m/z 158 and 174 for the structures without (14- and 15-membered macrocycles) and with a (16-membered macrocycle) glycosylated desosamine moiety, respectively. Their use as diagnostic ions in HPLC-MS/MS analysis in PrecIS detection mode allowed for the suspect screening of macrolides in municipal wastewater with the subsequent tentative identification and semi-quantitative determination of the detected compounds. The combination of this approach with preliminary solid-phase extraction provided analyte detection limits in the range of 4–150 ng L−1 and a linear range comprising at least two orders of magnitude. The application of the developed method to the study of the wastewater discharged into a natural reservoir after municipal treatment facilities made it possible to detect 17 compounds belonging to the macrolide class, including azithromycin (the dominant component), clarithromycin, josamycin and 14 metabolites with a total concentration of 1450 ng L−1. The observed level of macrolides indicates the insufficient effectiveness of the applied wastewater treatment technologies and the possibility of developing the macrolide-antibiotics resistance genes in bacteria. The developed method can be considered complementary to an HPLC-MS/MS in the MRM detection mode and can be implemented on the same equipment. It allows for the non-targeted screening of antibiotics and their metabolites, while MRM ensures their highly sensitive quantification if the appropriate analytical standards are available.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemosensors11010044/s1, Figure S1: Collision-induced dissociation pathways of erythromycin; Figure S2: Collision-induced dissociation pathways of clarithromycin; Figure S3: Collision-induced dissociation pathways of azithromycin; Figure S4: Collision-induced dissociation pathways of midecamycin; Figure S5: Collision-induced dissociation pathways of spiramycin; Figure S6: Collision-induced dissociation pathways of josamycin; Figure S7: HPLC-MS/MS (PrecIS) chromatogram of the model mixture macrolides with concentrations close to LOQ (diagnostic ions: m/z 174 (red) and m/z 158 (black)); Figure S8: Extracted ion current HPLC-HRMS chromatograms of municipal wastewater (peak numbers and m/z values correspond to those listed in Figure 4 and Table 4); Table S1: Recoveries of the macrolides obtained by solid-phase extraction on an HLB stationary phase; Table S2: Tandem mass spectra of the precursor ion candidates found in HPLC-MS/MS (PrecIS) chromatogram of municipal wastewater (compound numbers correspond to the peaks on Figure 4 and in Table 4).
Author Contributions
Conceptualization, I.S.V., D.I.F. and D.S.K.; methodology, I.S.V. and N.V.U.; validation, I.S.V. and N.V.U.; formal analysis, I.S.V. and D.I.F.; investigation, I.S.V.; resources, N.V.U.; data curation, I.S.V. and D.I.F.; writing—original draft preparation, I.S.V.; writing—review and editing, D.S.K.; visualization, I.S.V. and D.I.F.; supervision, D.S.K.; funding acquisition, D.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation, state assignment project number 0793-2020-0007.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained in the article and Supplementary Materials.
Acknowledgments
This study was performed using instrumentation of the Core Facility Center “Arktika” of the Lomonosov Northern (Arctic) Federal University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ye, X.; Sikirica, V.; Schein, J.R.; Grant, R.; Zarotsky, V.; Doshi, D.; Benson, C.J.; Riede, A.A. Treatment Failure Rates and Health Care Utilization and Costs among Patients with Community-Acquired Pneumonia Treated with Levofloxacin or Macrolides. in an Outpatient Setting: A Retrospective Claims Database Analysis. Clin. Ther. 2008, 30, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Culić, V.; Eraković, M.J. Parnham. Anti-inflammatory effects of macrolide antibiotics. Eur. J. Pharmcol. 2001, 429, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Gandra, S.; Ashok, A.; Caudron, Q.; Grenfell, B.T.; Levin, S.A.; Laxminarayan, R. Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet Infect. Dis. 2014, 14, 742–750. [Google Scholar] [CrossRef]
- World Health Organization. WHO Report on Surveillance of Antibiotic Consumption: 2016–2018 Early Implementation; WHO: Geneva, Switzerland, 2018; pp. 1–128. [Google Scholar]
- European Centre for Disease Prevention and Control. Antimicrobial Consumption. Annual Epidemiological Report for 2016; ECDC: Stockholm, Sweden, 2018; pp. 1–16. [Google Scholar]
- Grau, S.; Echeverria-Esnal, D.; Gómez-Zorrilla, S.; Navarrete-Rouco, M.E.; Masclans, J.R.; Espona, M.; Gracia-Arnillas, M.P.; Duran, X.; Comas, M.; Horcajada, J.P.; et al. Evolution of Antimicrobial Consumption during the First Wave of COVID-19. Pandemic. Antibiotics 2021, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Pani, A.; Lauriola, M.; Romandini, A.; Scaglione, F. Macrolides and viral infections: Focus on azithromycin in COVID-19 pathology. Int. J. Antimicrob. Agents 2020, 56, 106053. [Google Scholar] [CrossRef] [PubMed]
- Nippes, R.P.; Macruz, P.D.; da Silva, G.N.; Neves Olsen Scaliante, M.H. A critical review on environmental presence of pharmaceutical drugs tested for the COVID-19 treatment. Process Saf. Environ. Prot. 2021, 152, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Farooq, M.; Hanna, K. Environmental side effects of the injudicious use of antimicrobials in the era of COVID-19. Sci. Total Environ. 2020, 745, 141053. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Caarlsen, P.M.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [Google Scholar] [CrossRef]
- Wang, W.; Wang, H.; Zhang, W.; Liang, H.; Gao, D. Occurrence, distribution, and risk assessment of antibiotics in the Songhua River in China. Environ. Sci. Pollut. Res. Int. 2017, 24, 19282–19292. [Google Scholar] [CrossRef]
- Kümmerer, K. Drugs in the environment: Emission of drugs, diagnostic aids and disinfectants into wastewater by hospitals in relation to other sources—A review. Chemosphere 2001, 45, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Munir, M.; Xagoraraki, I. Correlation of tetracycline and sulfonamide antibiotics with corresponding resistance genes and resistant bacteria in a conventional municipal wastewater treatment plant. Sci. Total Environ. 2012, 421, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hu, X.; Xu, T.; Zhang, H.; Sheng, D.; Yi, D. Prevalence of antibiotic resistance genes and their relationship with antibiotics in the Huangpu River and the drinking water sources, Shanghai, China. Sci. Total Environ. 2013, 458, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, I.T.; Santos, L. Antibiotics in the aquatic environments: A review of the European scenario. Environ. Int. 2016, 94, 736–757. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Sherazi, S.T.H.; Mahesar, S.A.; Sirajuddin, M.A.; Malah, M.A. Brief overview of frequently used macrolides and analytical techniques for their assessment. Cur. Anal. Chem. 2019, 15, 324–338. [Google Scholar] [CrossRef]
- Ozumchelouei, J.E.; Hamidian, A.H.; Zhang, Y.; Yang, M. Physico-Chemical Properties of Antibiotics: A review with an emphasis on detection in the aquatic environment. Water Environ. Res. 2019, 92, 177–188. [Google Scholar] [CrossRef]
- Senta, I.; Terzic, S.; Ahel, M. Simultaneous Determination of Sulfonamides, Fluoroquinolones, Macrolides and Trimethoprim in Wastewater and River Water by LC-Tandem-MS. Chromatographia 2008, 68, 747–758. [Google Scholar] [CrossRef]
- Wang, Z.; Song, X.; Zhou, T.; Bian, K.; Zhang, F.; He, L. Simultaneous determination of ten macrolides drugs in feeds by high performance liquid chromatography with evaporation light scattering detection. RSC Adv. 2015, 5, 1491–1499. [Google Scholar] [CrossRef]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap. tandem mass spectrometry. J. Chromatog. A 2013, 1292, 173–188. [Google Scholar] [CrossRef]
- Benedetti, B.; Mauro, M.; Cavaliere, C.; Montone, C.M.; Fatone, F.; Frisonc, N.; Lagana, A.; Capriotti, A.L. Determination of multi-class emerging contaminants in sludge and recovery materials from waste water treatment plants: Development of a modified QuEChERS method coupled to LC–MS/MS. Microchem. J. 2020, 155, 104732. [Google Scholar] [CrossRef]
- Boix, C.; Ibáñez, M.; Sancho, J.V.; Rambla, J.; Aranda, J.L.; Ballester, S.; Hernandez, F. Fast determination of 40 drugs in water using large volume direct injection liquid chromatography–tandem mass spectrometry. Talanta 2015, 131, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xu, Y.; Sang, J.; Zhu, B.; Wang, J. Characterization of a new component and impurities in josamycin by trap-free two-dimensional liquid chromatography coupled to ion trap time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2019, 33, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zweigenbaum, J.A.; Thurman, E.M. Analysis of 70 Environmental Protection Agency priority pharmaceuticals in water by EPA Method 1694. J. Chromatog. A 2010, 1217, 5674–5686. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Skierska, J.; Szultka-Młyńska, M.; Pauter, K.; Buszewski, B. Study of chromatographic behavior of antibiotic drugs and their metabolites based on quantitative structure-retention relationships with the use of HPLC-DAD. J. Pharm. Biomed. Anal. 2020, 184, 113187. [Google Scholar] [CrossRef]
- Bichon, E.; Béasse, A.; Prevost, S.; Christien, S.; Courant, F.; Monteau, F.; Le Bizec, B. Improvement of estradiol esters monitoring in bovine hair by dansylation and liquid chromatography/tandem mass spectrometry analysis in multiple reaction. monitoring and precursor ion scan modes. Rapid Commun. Mass Spectrom. 2012, 26, 819–827. [Google Scholar] [CrossRef]
- Falev, D.I.; Ul’yanovskii, N.V.; Ovchinnikov, D.V.; Faleva, A.V.; Kosyakov, D.S. Screening and semi-quantitative determination of pentacyclic triterpenoids in plants by liquid chromatography–tandem mass spectrometry in precursor ion scan mode. Phytochem. Anal. 2020, 32, 252–261. [Google Scholar] [CrossRef]
- Zheng, F.; Xiao, H.M.; Zhu, Q.F.; Yu, Q.W.; Feng, Y.Q. Profiling of benzimidazoles and related metabolites in pig serum based on SiO2@NiO solid-phase extraction combined precursor ion scan with high resolution orbitrap mass spectrometry. Food Chem. 2019, 284, 279–286. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS; U.S. EPA: Washington, DC, USA, 2007; pp. 1–77. [Google Scholar]
- Mokh, S.; El Khatib, M.; Koubar, M.; Daher, Z.; Al Iskandarani, M. Innovative SPE-LC-MS/MS technique for the assessment of 63 pharmaceuticals and the detection of antibiotic-resistant-bacteria: A case study natural water sources in Lebanon. Sci. Total Environ. 2017, 609, 830–840. [Google Scholar] [CrossRef]
- Fohner, A.E.; Sparreboom, A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Macrolide antibiotic pathway, pharmacokinetics/pharmacodynamics. Pharmacogen. Genomics 2017, 27, 164–167. [Google Scholar] [CrossRef]
- Kibwage, I.O.; Janssen, G.; Busson, R.; Hoogmartens, J.; Vanderhaeghe, H.; Verbist, L. Identification of novel erythromycin derivatives in mother liquor concentrates of Streptomyces erythraeus. J. Antibiot. 1987, 40, 1–6. [Google Scholar] [CrossRef]
- Rodvold, K.A. Clinical Pharmacokinetics of Clarithromycin. Clin. Pharmacokinet. 1999, 37, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Weiss, K.; Maletzki, D.; Schüssler, W.; Schudoma, D.; Kopf, W.; Kühnen, U. Aquatic toxicity of the macrolide antibiotic clarithromycin and its metabolites. Chemosphere 2015, 120, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Gago-Ferrero, P.; Schymanski, E.L.; Bletsou, A.A.; Aalizadeh, R.; Hollender, J.; Thomaidis, N.S. Extended Suspect and Non-Target Strategies to Characterize Emerging Polar Organic Contaminants in Raw Wastewater with LC-HRMS/MS. Environ. Sci. Technol. 2015, 49, 12333–12341. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Dougherty, T.J.; Magee, T.V. Macrolide Antibiotics. Compr. Med. Chem. 2007, 7, 519–566. [Google Scholar] [CrossRef]
- Van den Bossche, L.; Daidone, F.; Van Schepdael, A.; Hoogmartens, J.; Adams, E. Characterization of impurities in josamycin using dual liquid chromatography combined with mass spectrometry. J. Pharm. Biomed. Anal. 2013, 73, 66–76. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Larsson, D.G.J. Concentrations of antibiotics predicted to select for resistant bacteria: Proposed limits for environmental regulation. Environ. Int. 2016, 86, 140–149. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).