
Composite Electrodes Based on Carbon Materials Decorated with Hg Nanoparticles for the Simultaneous Detection of Cd(II), Pb(II) and Cu(II)




Abstract
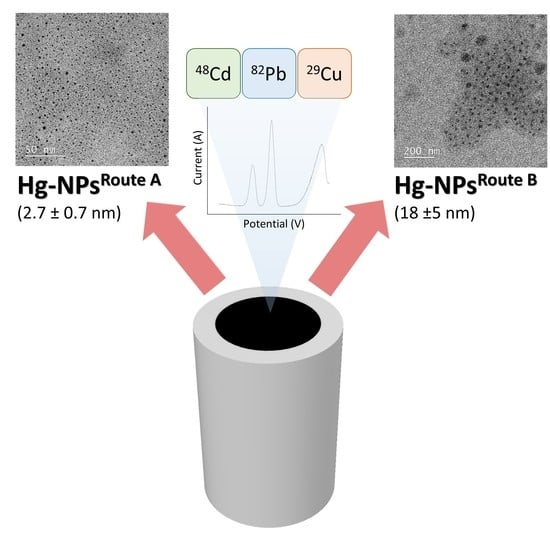
:
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Materials
2.2. Apparatus
2.3. Synthesis Hg-NPs
2.4. Electrode Fabrication
2.5. Drop-Casting Modification of Electrodes
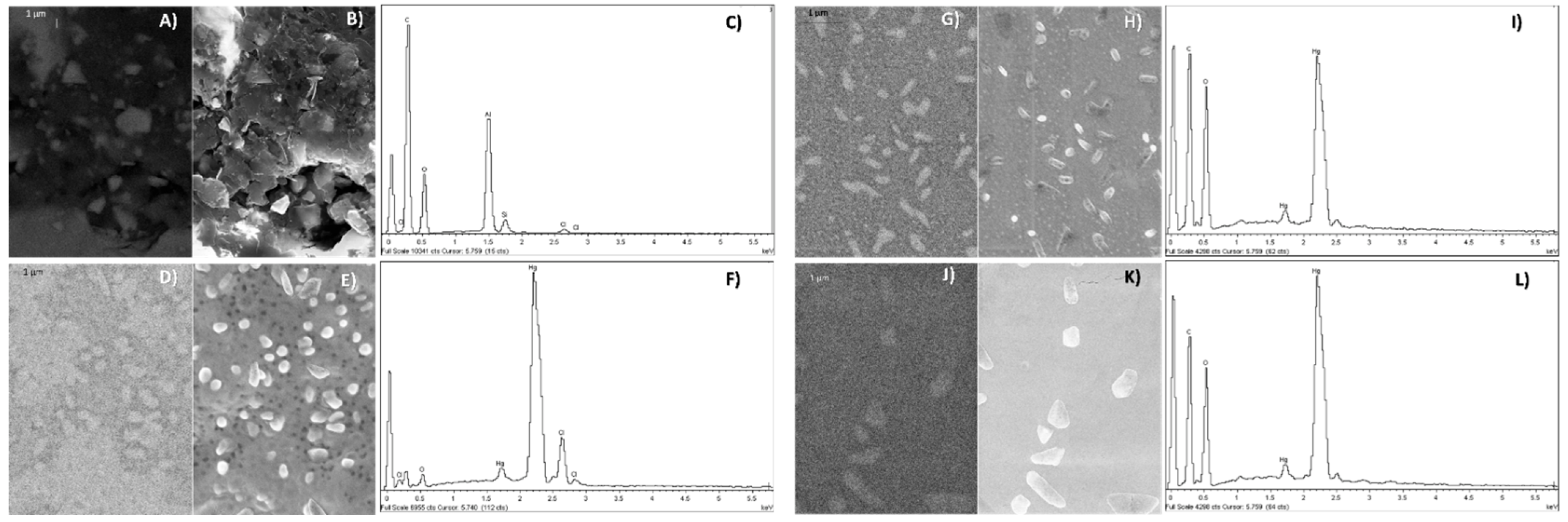
2.6. Morphological and Chemical Characterization
2.7. Electrochemical Characterization
2.8. Electroanalytical Measurements
3. Results
3.1. Morphological and Chemical Characterization of Hg-NPs
3.2. Electrochemical Characterization of Hg-NP-Modified Composite Electrodes
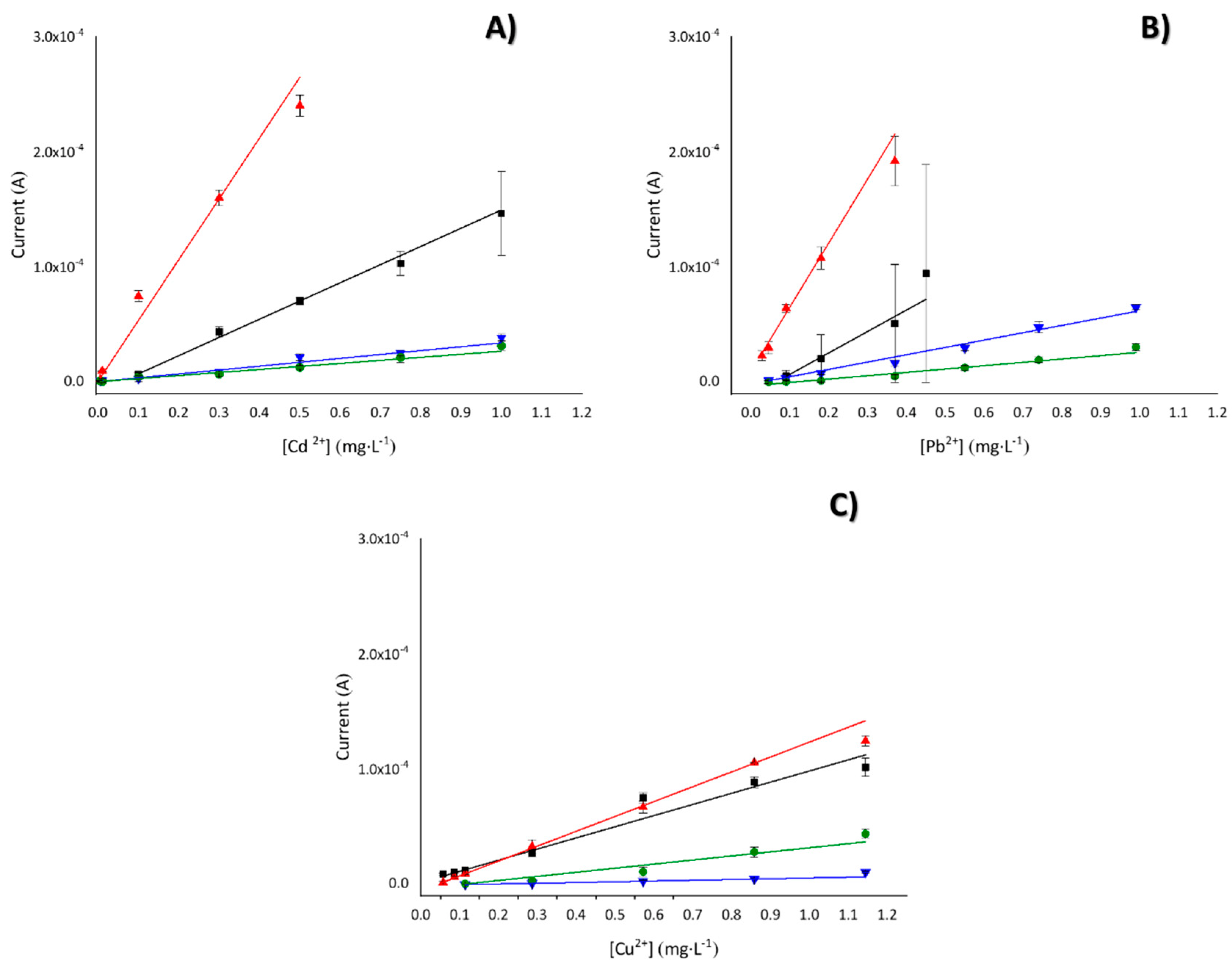
3.3. Analytical Characterization: Cation Metal Detection
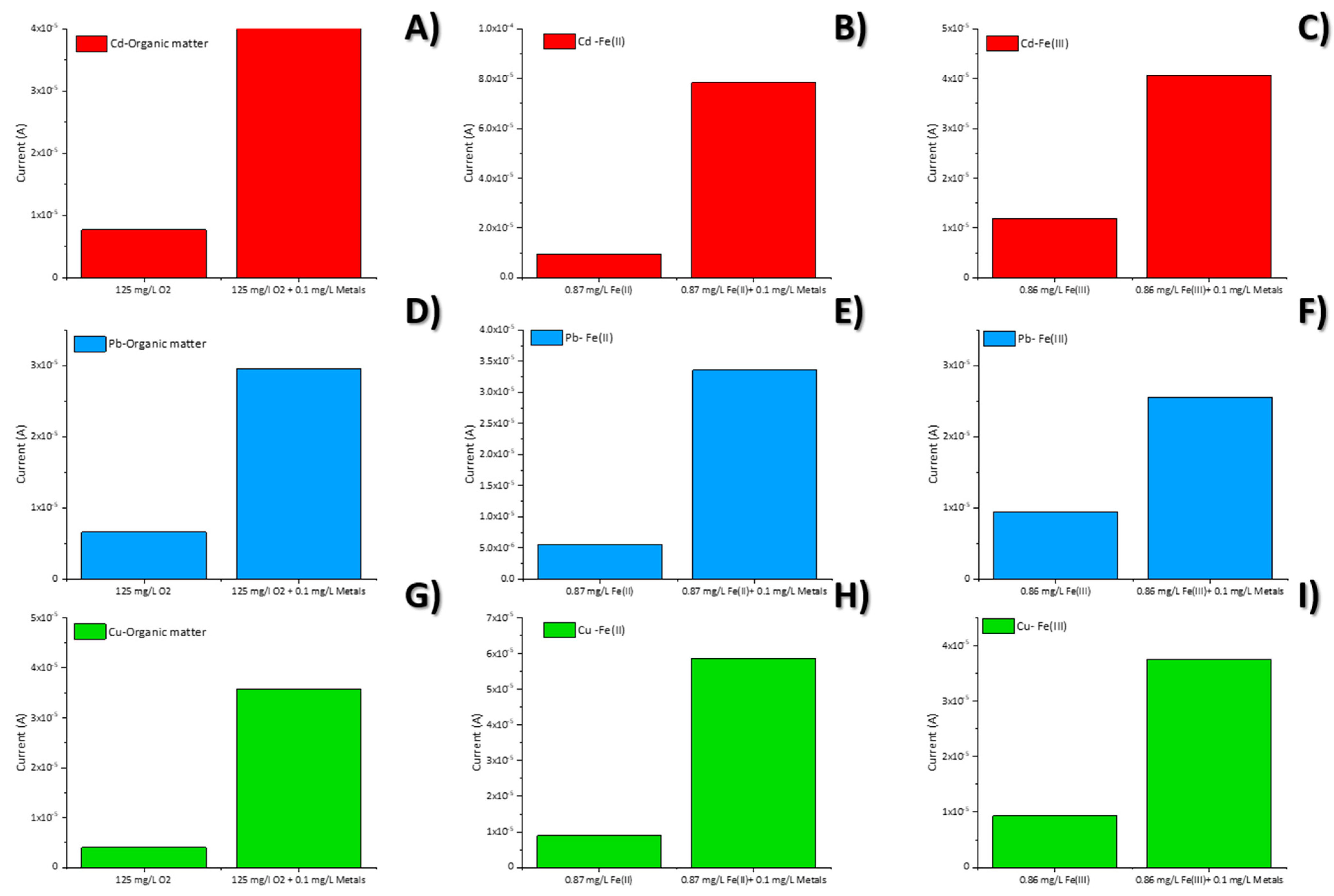
3.4. Interference Study
3.5. Suitability for Real Samples Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chubaka, C.E.; Whiley, H.; Edwards, J.W.; Ross, K.E. Lead, Zinc, Copper, and Cadmium Content of Water from South Australian Rainwater Tanks. Int. J. Environ. Res. Public Health 2018, 15, 1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaetke, L.M.; Chow, C.K. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 2003, 189, 147–163. [Google Scholar] [CrossRef]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Godt, J.; Scheidig, F.; Grosse-Siestrup, C.; Esche, V.; Brandenburg, P.; Reich, A.; A Groneberg, D. The toxicity of cadmium and resulting hazards for human health. J. Occup. Med. Toxicol. 2006, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, A.L.; Ara, A.; Usmani, J.A. Lead toxicity: A review. Interdiscip. Toxicol. 2015, 8, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Bocca, B.; Caimi, S.; Senofonte, O.; Alimonti, A.; Petrucci, F. ICP-MS based methods to characterize nanoparticles of TiO2 and ZnO in sunscreens with focus on regulatory and safety issues. Sci. Total Environ. 2018, 630, 922–930. [Google Scholar] [CrossRef]
- Lim, J.Y.; Mubarak, N.; Abdullah, E.C.; Nizamuddin, S.; Khalid, M. Inamuddin Recent trends in the synthesis of graphene and graphene oxide based nanomaterials for removal of heavy metals—A review. J. Ind. Eng. Chem. 2018, 66, 29–44. [Google Scholar] [CrossRef]
- Tseng, W.-C.; Hsu, K.-C.; Shiea, C.S.; Huang, Y.-L. Recent trends in nanomaterial-based microanalytical systems for the speciation of trace elements: A critical review. Anal. Chim. Acta 2015, 884, 1–18. [Google Scholar] [CrossRef]
- Bansod, B.K.; Kumar, T.; Thakur, R.; Rana, S.; Singh, I. A review on various electrochemical techniques for heavy metal ions detection with different sensing platforms. Biosens. Bioelectron. 2017, 94, 443–455. [Google Scholar] [CrossRef]
- Skoog, D.A.; James Holler, F.; Nieman, T.A. (Eds.) Principios de Análisis Instrumental, 5th ed.; McGrawHill: New York, NY, USA, 2001; 856p. [Google Scholar]
- Yao, Y.; Wu, H.; Ping, J. Simultaneous determination of Cd(II) and Pb(II) ions in honey and milk samples using a single-walled carbon nanohorns modified screen-printed electrochemical sensor. Food Chem. 2019, 274, 8–15. [Google Scholar] [CrossRef]
- Armstrong, K.C.; Tatum, C.E.; Dansby-Sparks, R.N.; Chambers, J.Q.; Xue, Z.-L. Individual and simultaneous determination of lead, cadmium, and zinc by anodic stripping voltammetry at a bismuth bulk electrode. Talanta 2010, 82, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, S.; Strack, G.; Kurup, P. Gold nanostar electrodes for heavy metal detection. Sens. Actuators B Chem. 2019, 281, 383–391. [Google Scholar] [CrossRef]
- Niu, P.; Fernández-Sánchez, C.; Gich, M.; Navarro-Hernández, C.; Fanjul-Bolado, P.; Roig, A. Screen-printed electrodes made of a bismuth nanoparticle porous carbon nanocomposite applied to the determination of heavy metal ions. Mikrochim. Acta 2016, 183, 617–623. [Google Scholar] [CrossRef]
- Ross, J.W.; DeMars, R.D.; Shain, I. Analytical Applications of Hanging Mercury Drop Electrode. Anal. Chem. 1956, 28, 1768–1771. [Google Scholar] [CrossRef]
- Pujol, L.; Evrard, D.; Serrano, K.G.; Freyssinier, M.; Ruffien-Cizsak, A.; Gros, P. Electrochemical sensors and devices for heavy metals assay in water: The French groups’ contribution. Front. Chem. 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Ariño, C.; Serrano, N.; Díaz-Cruz, J.M.; Esteban, M. Voltammetric determination of metal ions beyond mercury electrodes. A review. Anal. Chim. Acta 2017, 990, 11–53. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.; Montes, R.; Baeza, M. Trends in electrochemical impedance spectroscopy involving nanocomposite transducers: Characterization, architecture surface and bio-sensing. TrAC Trends Anal. Chem. 2017, 97, 201–215. [Google Scholar] [CrossRef]
- King, W.I. The Annals of Mathematical Statistics. Ann. Math. Stat. 1930, 1, 1–2. [Google Scholar] [CrossRef]
- Montes, R.; Bartrolí, J.; Céspedes, F.; Baeza, M. Towards to the improvement of the analytical response in voltammetric sensors based on rigid composites. J. Electroanal. Chem. 2014, 733, 69–76. [Google Scholar] [CrossRef]
- Chaiyo, S.; Mehmeti, E.; Žagar, K.; Siangproh, W.; Chailapakul, O.; Kalcher, K. Electrochemical sensors for the simultaneous determination of zinc, cadmium and lead using a Nafion/ionic liquid/graphene composite modified screen-printed carbon electrode. Anal. Chim. Acta 2016, 918, 26–34. [Google Scholar] [CrossRef]
- Truzzi, C.; Lambertucci, L.; Gambini, G.; Scarponi, G. Optimization of square wave anodic stripping voltammetry (SWASV) for the simultaneous determination of Cd, Pb, and Cu in seawater and comparison with differential pulse anodic stripping voltammetry (DPASV). Ann. Chim. 2002, 92, 313–326. [Google Scholar] [PubMed]
- Muñoz, C.; Zuñiga, M.; Arancibia, V. Stripping voltammetric determination of cadmium in sea water using a carbon paste electrode modified with alginic acid from brown algae. J. Braz. Chem. Soc. 2010, 21, 1688–1691. [Google Scholar] [CrossRef] [Green Version]
- Englich, F.V.; Foo, T.C.; Richardson, A.C.; Ebendorff-Heidepriem, H.; Sumby, C.J.; Monro, T.M. Photoinduced Electron Transfer Based Ion Sensing within an Optical Fiber. Sensors 2011, 11, 9560–9572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragay, G.; Pons, J.; Merkoçi, A. Recent Trends in Macro-, Micro-, and Nanomaterial-Based Tools and Strategies for Heavy-Metal Detection. Chem. Rev. 2011, 111, 3433–3458. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Karim, R.; Reda, Y.; Abdel-Fattah, A. Review—Nanostructured Materials-Based Nanosensors. J. Electrochem. Soc. 2020, 167, 037554. [Google Scholar] [CrossRef]
- Hong, N.H. Introduction to Nanomaterials: Basic Properties, Synthesis, and Characterization; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Bastos-Arrieta, J.; Montes, R.; Ocaña, C.; Espinoza, M.; Muñoz, M.; Baeza, M. In situ characterization of size, spatial distribution, chemical composition, and electroanalytical response of hybrid nanocomposite materials. In In-Situ Characterization Techniques for Nanomaterials; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Ajitha, B.; Reddy, Y.A.K.; Reddy, P.S.; Jeon, H.-J.; Ahn, C.W. Role of capping agents in controlling silver nanoparticles size, antibacterial activity and potential application as optical hydrogen peroxide sensor. RSC Adv. 2016, 6, 36171–36179. [Google Scholar] [CrossRef]
- Regnery, S.; Ehrhart, P.; Szot, K.; Waser, R.; Ding, Y.; Jia, C.L.; Schumacher, M.; Mcentee, T. MOCVD of (Ba,Sr)TiO3: Nucleation and Growth. Integr. Ferroelectr. 2003, 57, 1175–1184. [Google Scholar] [CrossRef]
- Ramesh, G.V.; Prasad, M.D.; Radhakrishnan, T.P. Mercury Nanodrops and Nanocrystals. Chem. Mater. 2011, 23, 5231–5236. [Google Scholar] [CrossRef]
- Dong, L.; Zhang, X.; Ren, S.; Lei, T.; Sun, X.; Qi, Y.; Wu, Q. Poly(diallyldimethylammonium chloride)-cellulose nanocrystals supported Au nanoparticles for nonenzymatic glucose sensing. RSC Adv. 2016, 6, 6436–6442. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Jiang, S.P. Self-assembly of mixed Pt and Au nanoparticles on PDDA-functionalized graphene as effective electrocatalysts for formic acid oxidation of fuel cells. Phys. Chem. Chem. Phys. 2011, 13, 6883–6891. [Google Scholar] [CrossRef]
- Torres-Rivero, K.; Torralba-Cadena, L.; Espriu-Gascon, A.; Casas, I.; Bastos-Arrieta, J.; Florido, A. Strategies for Surface Modification with Ag-Shaped Nanoparticles: Electrocatalytic Enhancement of Screen-Printed Electrodes for the Detection of Heavy Metals. Sensors 2019, 19, 4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ref, L. Determinación de Cobre en Agua. Available online: https://www.dropsens.com/pdfs_productos/new_brochures/pl1_pl2_pl3_pl4_pl5_pl6_pl7.pdf (accessed on 13 September 2020).
- Landon, P.B.; Mo, A.H.; Zhang, C.; Emerson, C.D.; Printz, A.; Gomez, A.F.; Delatorre, C.J.; Colburn, D.A.M.; Anzenberg, P.; Eliceiri, M.; et al. Designing Hollow Nano Gold Golf Balls. ACS Appl. Mater. Interfaces 2014, 6, 9937–9941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shen, W.; Xue, J.; Liu, Y.; Liu, Y.; Yan, P.; Liu, J.; Tang, J. Recent advances in synthetic methods and applications of silver nanostructures. Nanoscale Res. Lett. 2018, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowiec, J.; Yan, K.; Tin, C.-C.; Zhang, J. Synthesis of PDDA Functionalized Reduced Graphene Oxide Decorated with Gold Nanoparticles and Its Electrochemical Response toward Levofloxacin. J. Electrochem. Soc. 2014, 162, H164–H169. [Google Scholar] [CrossRef]
- Yu, C.-J.; Lin, C.-Y.; Liu, C.-H.; Cheng, T.-L.; Tseng, W.-L. Synthesis of poly(diallyldimethylammonium chloride)-coated Fe3O4 nanoparticles for colorimetric sensing of glucose and selective extraction of thiol. Biosens. Bioelectron. 2010, 26, 913–917. [Google Scholar] [CrossRef]
- Fairley, N.; Fernandez, V.; Richard-Plouet, M.; Guillot-Deudon, C.; Walton, J.; Smith, E.; Flahaut, D.; Greiner, M.; Biesinger, M.; Tougaard, S.; et al. Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy. Appl. Surf. Sci. Adv. 2021, 5, 100112. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. NIST X-ray Photoelectron Spectroscopy Database. In NIST Standard Reference Database Number 20; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2000; 20899. Available online: https://srdata.nist.gov/xps/Default.aspx (accessed on 27 February 2022).
- Briggs, D. X-ray photoelectron spectroscopy (XPS). In Handbook of Adhesion, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 621–622. [Google Scholar] [CrossRef]
- Rudnicki, K.; Brycht, M.; Leniart, A.; Domagała, S.; Kaczmarek, K.; Kalcher, K.; Skrzypek, S. A Sensitive Sensor Based on Single-walled Carbon Nanotubes: Its Preparation, Characterization and Application in the Electrochemical Determination of Drug Clorsulon in Milk Samples. Electroanalysis 2019, 32, 375–383. [Google Scholar] [CrossRef]
- Chakrabarti, D.J.; Laughlin, D.E. The Cu-Hg (Copper-Mercury) system. Bull. Alloy Phase Diagr. 1985, 6, 522–527. [Google Scholar] [CrossRef]


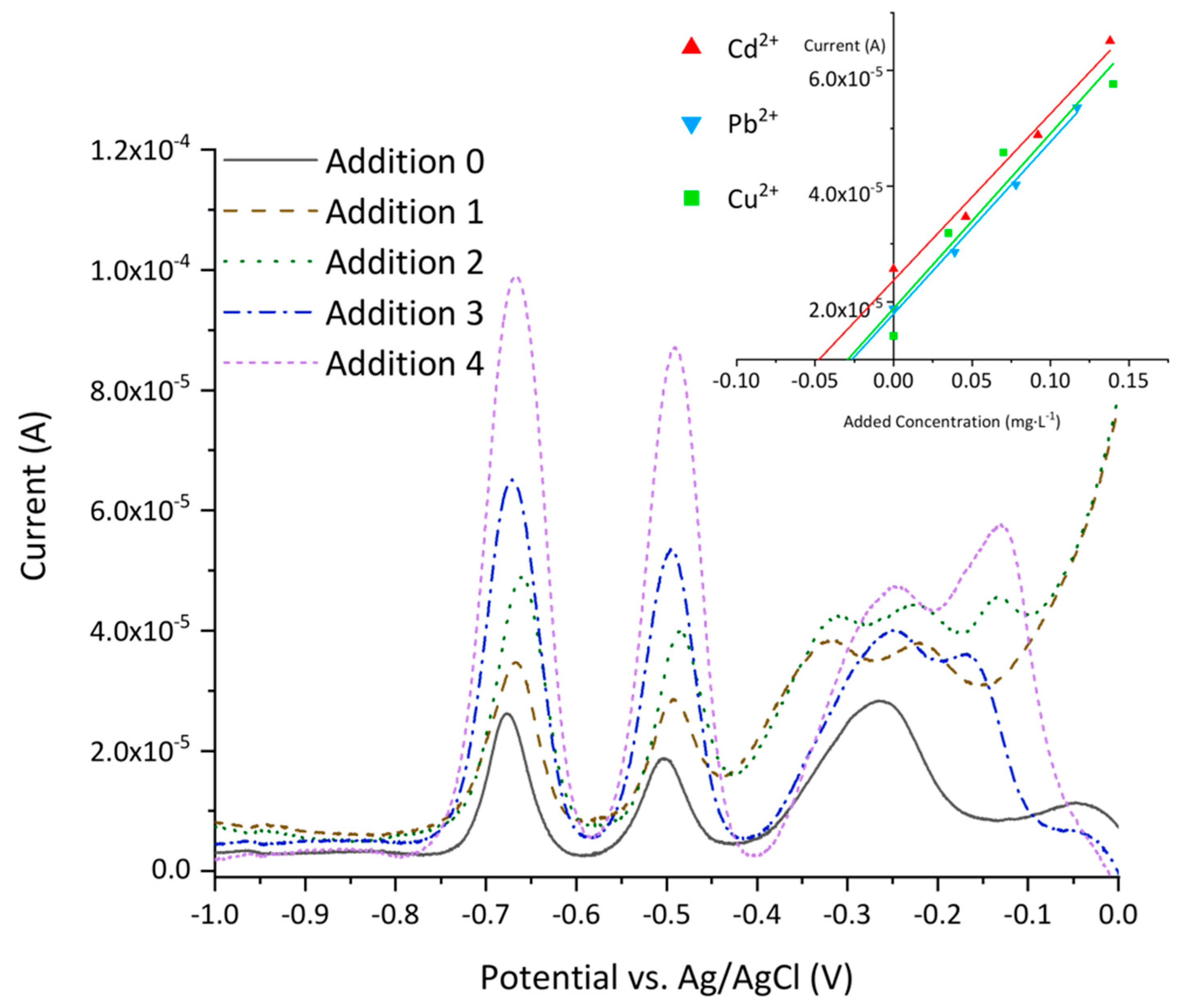

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cd2+ | |||
|---|---|---|---|
| Electrode | Sensitivity [A·L·mg−1] | R2 (n = 5) | Linear Range [mg·L−1] |
| Bare | (1.6 ± 0.1) × 10−4 | 0.995 | 0.10–1.0 |
| Hg-NPsRoute A | (5.3 ± 0.7) × 10−4 | 0.93 | 0.0050–0.50 |
| Hg-NPsRoute B | (3.4 ± 0.2) × 10−5 | 0.98 | 0.050–1.0 |
| Hg-NPsRoute B/PDDA | (2.6 ± 0.3) × 10−5 | 0.94 | 0.050–1.0 |
| Pb2+ | |||
| Electrode | Sensitivity [A·L·mg−1] | R2 (n = 5) | Linear Range [mg·L−1] |
| Bare | (1.9 ± 0.2) × 10−4 | 0.95 | 0.090–0.45 |
| Hg-NPsRoute A | (5.6 ± 0.5) × 10−4 | 0.96 | 0.028–0.37 |
| Hg-NPsRoute B | (6.4 ± 0.3) × 10−5 | 0.98 | 0.045–1.0 |
| Hg-NPsRoute B/PDDA | (2.9 ± 0.3) × 10−5 | 0.93 | 0.045–1.0 |
| Cu2+ | |||
| Electrode | Sensitivity [A·L·mg−1] | R2 (n = 5) | Linear Range [mg·L−1] |
| Bare | (9.7 ± 0.9) × 10−5 | 0.95 | 0.057–1.1 |
| Hg-NPsRoute A | (1.29 ± 0.02) × 10−4 | 0.999 | 0.057–1.1 |
| Hg-NPsRoute B | (7 ± 1) × 10−6 | 0.90 | 0.11–1.1 |
| Hg-NPsRoute B/PDDA | (3.6 ± 0.6) × 10−5 | 0.90 | 0.11–1.1 |
| Metal | Sensitivity [A·L·mg−1] | R2 | Linear Range [mg·L−1] | DL [mg·L−1] |
|---|---|---|---|---|
| Cd2+ | (8.4 ± 0.6) × 10−4 | 0.99 (n = 4) | 0.0050–0.30 | 0.0015 |
| Pb2+ | (8 ± 1) × 10−4 | 0.95 (n = 4) | 0.046–0.37 | 0.02 |
| Cu2+ | (2.1 ± 0.3) × 10−4 | 0.95 (n = 4) | 0.086–0.57 | 0.057 |
| Metal | Reproducibility [A·L·mg−1] (n = 3) | Repeatability [A·L·mg−1] (n = 2) |
|---|---|---|
| Cd2+ | (5 ± 1) × 10−4 | (4 ± 1) × 10−4 |
| Pb2+ | (6 ± 1) × 10−4 | (4 ± 1) × 10−4 |
| Cu2+ | (2 ± 1) × 10−4 | (8 ± 1) × 10−5 |
| Metal | Hg-NPsRoute A Electrode [mg·L−1] | ICP-MS [mg·L−1] | Recovery (%) |
|---|---|---|---|
| Cd2+ | 2.06 | 1.9 | 108% |
| Pb2+ | 1.51 | 1.6 | 94% |
| Cu2+ | 1.56 | 1.5 | 104% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández, L.L.; Bastos-Arrieta, J.; Palet, C.; Baeza, M. Composite Electrodes Based on Carbon Materials Decorated with Hg Nanoparticles for the Simultaneous Detection of Cd(II), Pb(II) and Cu(II). Chemosensors 2022, 10, 148. https://doi.org/10.3390/chemosensors10040148
Fernández LL, Bastos-Arrieta J, Palet C, Baeza M. Composite Electrodes Based on Carbon Materials Decorated with Hg Nanoparticles for the Simultaneous Detection of Cd(II), Pb(II) and Cu(II). Chemosensors. 2022; 10(4):148. https://doi.org/10.3390/chemosensors10040148
Chicago/Turabian StyleFernández, Laia L., Julio Bastos-Arrieta, Cristina Palet, and Mireia Baeza. 2022. "Composite Electrodes Based on Carbon Materials Decorated with Hg Nanoparticles for the Simultaneous Detection of Cd(II), Pb(II) and Cu(II)" Chemosensors 10, no. 4: 148. https://doi.org/10.3390/chemosensors10040148
APA StyleFernández, L. L., Bastos-Arrieta, J., Palet, C., & Baeza, M. (2022). Composite Electrodes Based on Carbon Materials Decorated with Hg Nanoparticles for the Simultaneous Detection of Cd(II), Pb(II) and Cu(II). Chemosensors, 10(4), 148. https://doi.org/10.3390/chemosensors10040148