


Lost in Transition: Challenges in the Journey from Pediatric to Adult Care for a Romanian DMD Patient

 ,
,  and
and 
Abstract
1. Introduction
2. Case Presentation
2.1. Methodology
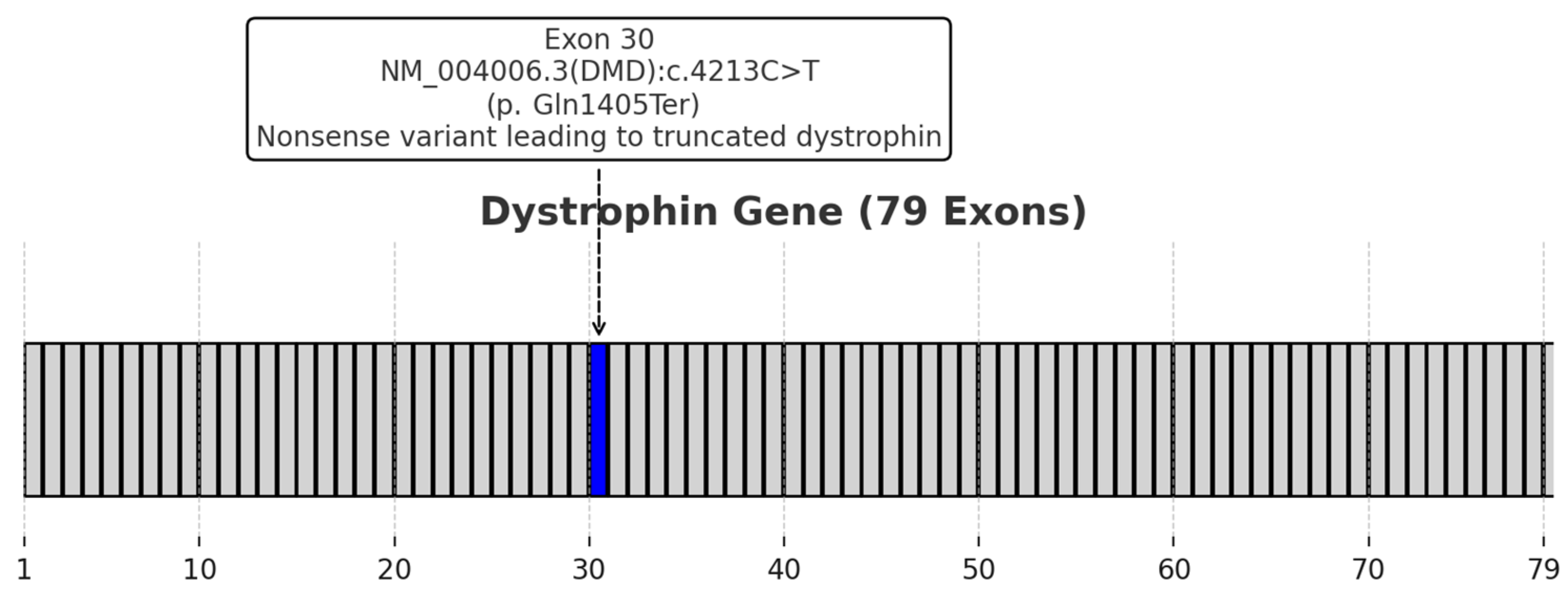
2.2. Clinical Presentation and Diagnosis
2.3. Multidisciplinary Management
2.4. Transition Process
2.5. Patient Perspective
“I find managing my medication on my own quite challenging. My mother handles everything related to prescriptions, purchasing, and managing potential side effects. It’s simply not something I can manage independently. Scheduling and attending appointments have become more difficult since transitioning from pediatric to adult care. While I don’t face issues with transportation, as we have a personal car, organizing consultations and ensuring all necessary tests are completed can still be complicated.When speaking with doctors, I feel confident in their expertise and explanations. If I don’t fully understand something, I always ask for additional clarification, making sure it’s explained in a way that I can grasp. I feel very involved in decisions regarding my health, but I’m not confident I could manage everything on my own. This is mainly due to the mobility challenges I face, which make many aspects of daily life, including healthcare, hard to navigate independently.The biggest difficulty I’ve encountered is finding the right specialists for my condition. To make the transition smoother, I think there should be dedicated teams of specialists who are well-informed about Duchenne Muscular Dystrophy and able to provide comprehensive care for patients like me.These insights reflect my experiences and highlight the importance of creating a better, more supportive transition process for patients with DMD.”
3. Discussion
3.1. Multidisciplinary Team
3.2. Challenges and Opportunities in Transition Care for Romanian DMD Patients
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Wasilewska, E.; Małgorzewicz, S.; Sobierajska-Rek, A.; Jabłońska-Brudło, J.; Górska, L.; Śledzińska, K.; Bautembach-Minkowska, J.; Wierzba, J. Transition from childhood to adulthood in patients with duchenne muscular dystrophy. Medicina 2020, 56, 426. [Google Scholar] [CrossRef] [PubMed]
- Van Ruiten, H.; Bushby, K.; Guglieri, M. State-of-the-art advances in duchenne muscular dystrophy. EMJ Eur. Med. J. 2017, 2, 90–99. [Google Scholar]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Lupu, M.; Mihaela Pintilie, I.; Teleanu, R.I.; Marin, G.G.; Vladâcenco, O.A.; Severin, E.M. Early Cardiac Dysfunction in Duchenne Muscular Dystrophy: A Case Report and Literature Update. Int. J. Mol. Sci. 2025, 26, 1685. [Google Scholar] [CrossRef]
- Castro, D.; Sejersen, T.; Bello, L.; Buccella, F.; Cairns, A.; Carranza-del Río, J.; de Groot, I.J.; Elman, L.; Inzani, I.; Klein, A.; et al. Transition of patients with Duchenne muscular dystrophy from paediatric to adult care: An international Delphi consensus study. Eur. J. Paediatr. Neurol. 2025, 54, 130–139. [Google Scholar]
- Lupu, M.; Ioghen, M.; Perjoc, R.Ș.; Scarlat, A.M.; Vladâcenco, O.A.; Roza, E.; Epure, D.A.-M.; Teleanu, R.I.; Severin, E.M. The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases. Children 2023, 10, 959. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Fleischer, M.; Fleischer, M.; Coskun, B.; Coskun, B.; Stolte, B.; Stolte, B.; Della-Marina, A.; Della-Marina, A.; Kölbel, H.; Kölbel, H.; et al. Essen transition model for neuromuscular diseases. Neurol. Res. Pract. 2022, 4, 41. [Google Scholar] [CrossRef]
- TTrout, C.J.; Case, L.E.; Clemens, P.R.; McArthur, A.; Noritz, G.; Ritzo, M.; Wagner, K.R.; Vroom, E.; Kennedy, A. A Transition Toolkit for Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S110–S117. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren delays loss of ambulation and respiratory decline in nonsense mutation Duchenne muscular dystrophy patients. J. Comp. Eff. Res. 2022, 11, 139–155. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Torella, A.; Zanobio, M.; Zeuli, R.; Del Vecchio Blanco, F.; Savarese, M.; Giugliano, T.; Garofalo, A.; Piluso, G.; Politano, L.; Nigro, V. The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS ONE 2020, 15, e0237803. [Google Scholar] [CrossRef]
- Zhou, H.; Fu, M.; Mao, B.; Yuan, L. Cardiac Phenotype–Genotype Associations in DMD/BMD: A Meta-Analysis and Systematic Review. Pediatr. Cardiol. 2021, 42, 189–198. [Google Scholar] [CrossRef]
- Harada, Y.; Sorensen, S.T.; Aravindhan, A.; Stefans, V.; Veerapandiyan, A. Dystrophinopathy in a Family Due to a Rare Nonsense Mutation Causing Predominant Behavioral Phenotype. J. Pediatr. Neurol. 2020, 18, 210–213. [Google Scholar] [CrossRef]
- Mukherjee, S.; Roy, M.; Guha, G.; Saha, S.P. Mutation location and cognitive impairment in duchenne muscular dystrophy. J. Neurosci. Rural. Pract. 2018, 9, 410–413. [Google Scholar] [CrossRef]
- Brogna, C.; Coratti, G.; Rossi, R.; Neri, M.; Messina, S.; Amico, A.D.; Bruno, C.; Lucibello, S.; Vita, G.; Berardinelli, A.; et al. The nonsense mutation stop+4 model correlates with motor changes in Duchenne muscular dystrophy. Neuromuscul. Disord. 2021, 31, 479–488. [Google Scholar] [CrossRef]
- MacKintosh, E.W.; Chen, M.L.; Benditt, J.O. Lifetime Care of Duchenne Muscular Dystrophy. Sleep Med. Clin. 2020, 15, 485–495. [Google Scholar] [CrossRef]
- Wood, D.L.; Sawicki, G.S.; Miller, M.D.; Smotherman, C.; Lukens-Bull, K.; Livingood, W.C.; Ferris, M.; Kraemer, D.F. The Transition Readiness Assessment Questionnaire (TRAQ): Its factor structure, reliability, and validity. Acad. Pediatr. 2014, 14, 415–422. [Google Scholar] [PubMed]
- Blum, R.W.; Garell, D.; Hodgman, C.H.; Jorissen, T.W.; Okinow, N.A.; Orr, D.P.; Slap, G.B. Transition from child-centered to adult health-care systems for adolescents with chronic conditions. A position paper of the Society for Adolescent Medicine. J. Adolesc. Health 1993, 14, 570–576. [Google Scholar] [PubMed]
- Callahan, S.T.; Feinstein Winitzer, R.; Keenan, P. Transition from pediatric to adult-oriented health care: A challenge for patients with chronic disease. Curr. Opin. Pediatr. 2001, 13, 310–316. [Google Scholar]
- Saito, T.; Kawai, M.; Kimura, E.; Ogata, K.; Takahashi, T.; Kobayashi, M.; Takada, H.; Kuru, S.; Mikata, T.; Matsumura, T.; et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul. Disord. 2017, 27, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, Y.; Mistry, B.; Gibson, B.E. Transitioning to adulthood with a progressive condition: Best practice assumptions and individual experiences of young men with Duchenne muscular dystrophy. Disabil. Rehabil. 2015, 37, 1144–1151. [Google Scholar] [CrossRef]
- Rosen, D.S.; Blum, R.W.; Britto, M.; Sawyer, S.M.; Siegel, D.M. Transition to adult health care for adolescents and young adults with chronic conditions: Position paper of the Society for Adolescent Medicine. J. Adolesc. Health 2003, 33, 309–311. [Google Scholar] [CrossRef]
- Rodger, S.; Steffensen, B.F.; Lochmüller, H. Transition from childhood to adulthood in Duchenne muscular dystrophy (DMD). Orphanet J. Rare Dis. 2012, 7, A8. [Google Scholar] [CrossRef]
- Abbott, D. Other Voices, Other Rooms: Reflections on Talking to Young Men with Duchenne Muscular Dystrophy and Their Families About Transition to Adulthood. Child. Soc. 2012, 26, 241–250. [Google Scholar] [CrossRef]
- Cooley, W.C.; Sagerman, P.J.; American Academy of Pediatrics; American Academy of Family Physicians; American College of Physicians; Transitions Clinical Report Authoring Group. Clinical report—Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics 2011, 128, 182–200. [Google Scholar] [CrossRef]
- Verhaert, D.; Richards, K.; Rafael-Fortney, J.A.; Raman, S.V. Cardiac involvement in patients with muscular dystrophies magnetic resonance imaging phenotype and genotypic considerations. Circ. Cardiovasc. Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Ararat, E.; Mhanna, M.J. Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr. Pulmonol. 2016, 51, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, D.; Khan, S.; Khera, R.; Bhatt, A.; Garg, S.; Grodin, J.L.; Morlend, R.; Araj, F.G.; Amin, A.A.; Thibodeau, J.T.; et al. Predictors of death in adults with duchenne muscular dystrophy-associated cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e006340. [Google Scholar] [CrossRef]
- Case, L.E.; Apkon, S.D.; Eagle, M.; Gulyas, A.; Juel, L.; Matthews, D.; Newton, R.A.; Posselt, H.F. Rehabilitation Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S17–S33. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD mutations in 576 dystrophinopathy families: A step forward in genotype-phenotype correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, H. Duchenne muscular dystophy: A short review and treatment update. Iran. J. Child Neurol. 2021, 15, 9–15. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed]
- Hildyard, J.C.W.; Crawford, A.H.; Rawson, F.; Riddell, D.O.; Harron, R.C.M.; Piercy, R.J. Single-transcript multiplex in situ hybridisation reveals unique patterns of dystrophin isoform expression in the developing mammalian embryo. Wellcome Open Res. 2020, 5, 76. [Google Scholar] [CrossRef]
- Lidov, H.G.W. Dystrophin in the nervous system. Brain Pathol. 1996, 6, 63–77. [Google Scholar] [CrossRef]
- Vaillend, C.; Aoki, Y.; Mercuri, E.; Hendriksen, J.; Tetorou, K.; Goyenvalle, A.; Muntoni, F. Duchenne muscular dystrophy: Recent insights in brain related comorbidities. Nat. Commun. 2025, 16, 1298. [Google Scholar] [CrossRef]
- Teleanu, R.I. (Ed.) Esențialul în Neurologia Pediatrică; Universitară Carol Davila: București, Romania, 2022. [Google Scholar]
- Fujino, H.; Iwata, Y.; Saito, T.; Matsumura, T.; Fujimura, H.; Imura, O. The experiences of patients with Duchenne muscular dystrophy in facing and learning about their clinical conditions. Int. J. Qual. Stud. Health Well-Being 2016, 11, 32045. [Google Scholar] [CrossRef]
- Hendriksen, J.G.M.; Poysky, J.T.; Schrans, D.G.M.; Schouten, E.G.W.; Aldenkamp, A.P.; Vles, J.S.H. Psychosocial adjustment in males with Duchenne muscular dystrophy: Psychometric properties and clinical utility of a parent-report questionnaire. J. Pediatr. Psychol. 2009, 34, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Poysky, J. Behavior patterns in Duchenne muscular dystrophy: Report on the Parent Project Muscular Dystrophy behavior workshop 8–9 of December 2006, Philadelphia, USA. Neuromuscul. Disord. 2007, 17, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Translarna: EMA Re-Confirms Non-Renewal of Authorisation of Duchenne Muscular Dystrophy Medicine. Available online: https://www.ema.europa.eu/en/news/translarna-ema-re-confirms-non-renewal-authorisation-duchenne-muscular-dystrophy-medicine (accessed on 17 February 2025).

{kind=link}
| TRAQ Domain | Assessment | Findings |
|---|---|---|
| Medication Management | Moderate | Needed caregiver assistance for refilling prescriptions and recognizing adverse effects. |
| Appointment Adherence | Moderate | Required reminders for scheduling and attending medical visits. |
| Health Monitoring | Low to moderate | Needed guidance on recognizing symptoms that warrant medical attention. |
| Healthcare Communication | High | Effectively discussed medical needs with providers. |
| Self-Advocacy and Decision-Making | Moderate | Expressed some concerns about transitioning to independent healthcare. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lupu, M.; Marcu, M.-A.; Epure, D.A.; Vladacenco, O.A.; Severin, E.M.; Teleanu, R.I. Lost in Transition: Challenges in the Journey from Pediatric to Adult Care for a Romanian DMD Patient. Healthcare 2025, 13, 830. https://doi.org/10.3390/healthcare13070830
Lupu M, Marcu M-A, Epure DA, Vladacenco OA, Severin EM, Teleanu RI. Lost in Transition: Challenges in the Journey from Pediatric to Adult Care for a Romanian DMD Patient. Healthcare. 2025; 13(7):830. https://doi.org/10.3390/healthcare13070830
Chicago/Turabian StyleLupu, Maria, Maria-Alexandra Marcu, Diana Anamaria Epure, Oana Aurelia Vladacenco, Emilia Maria Severin, and Raluca Ioana Teleanu. 2025. "Lost in Transition: Challenges in the Journey from Pediatric to Adult Care for a Romanian DMD Patient" Healthcare 13, no. 7: 830. https://doi.org/10.3390/healthcare13070830
APA StyleLupu, M., Marcu, M.-A., Epure, D. A., Vladacenco, O. A., Severin, E. M., & Teleanu, R. I. (2025). Lost in Transition: Challenges in the Journey from Pediatric to Adult Care for a Romanian DMD Patient. Healthcare, 13(7), 830. https://doi.org/10.3390/healthcare13070830