A Backward Technique for Demographic Noise in Biological Ordinary Differential Equation Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
- In Section 2.1 we provide a background to the modeling of gene regulation, pointing out the reason why it is an intrinsically stochastic mechanism and describing the most common simulation techniques.
- In Section 2.2 we illustrate an example of modeling and simulation of a GRN in bacteriophage , based on the paper by Goutsias [20] and the simulation technique described in [17].
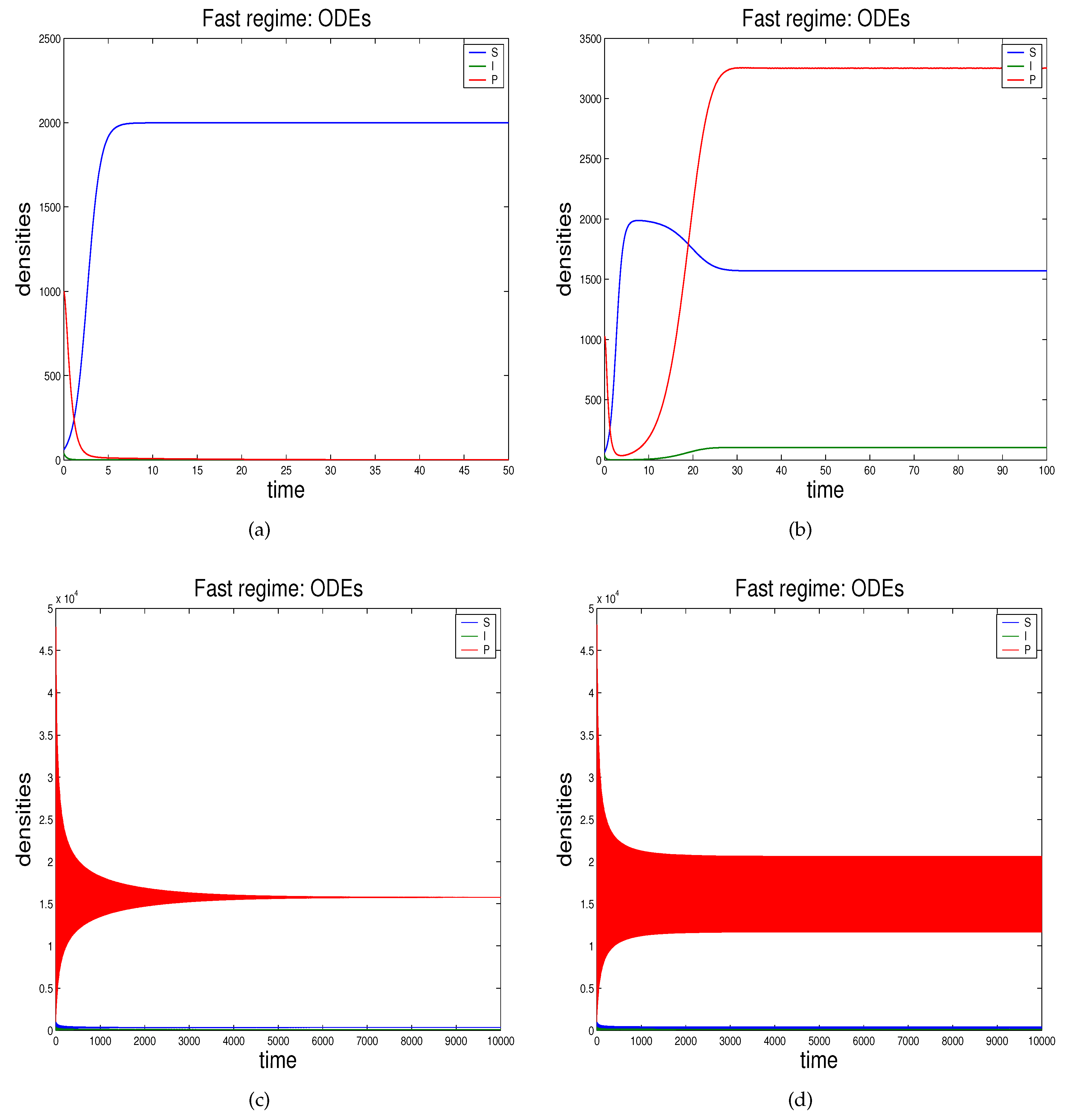
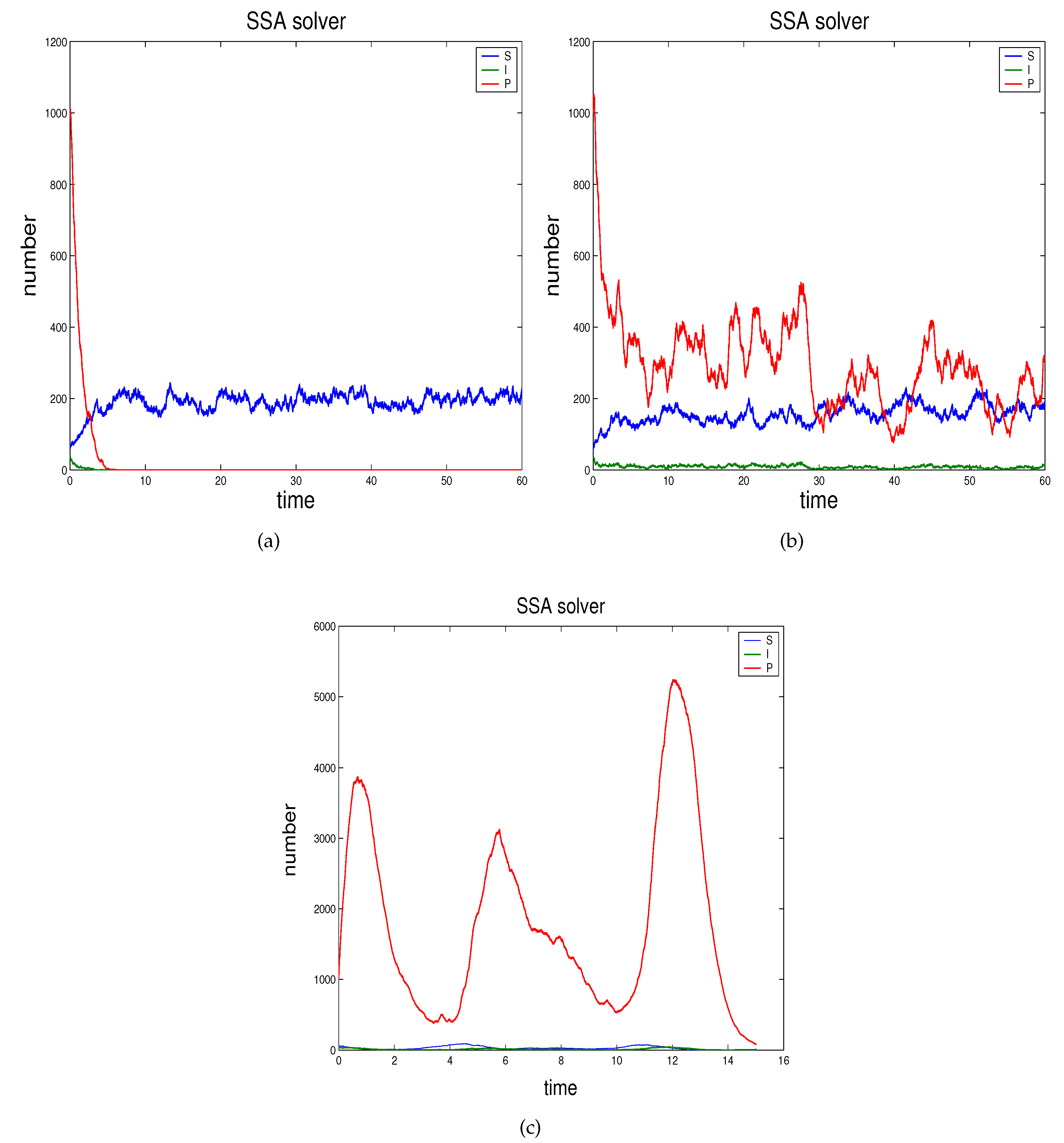
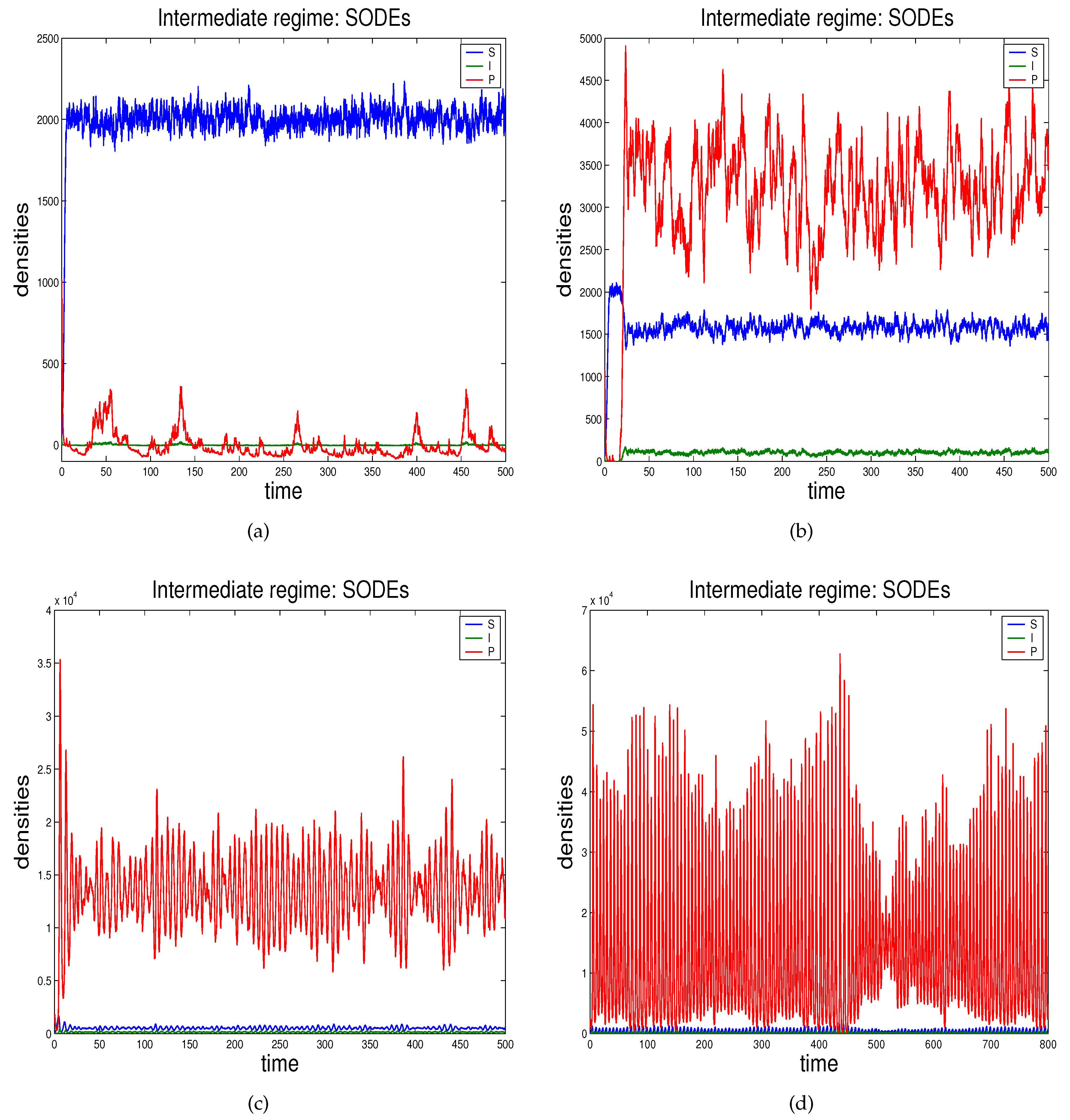
- In Section 2.3 we propose the novel technique through which we can study the effects of demographic noise in the ODEs model by Beretta and Kuang [16], going backwards to the discrete stochastic process and the continuous stochastic system (characterised by SODEs) underlying the deterministic model.
2.1. Stochastic Modeling of Genetic Regulation
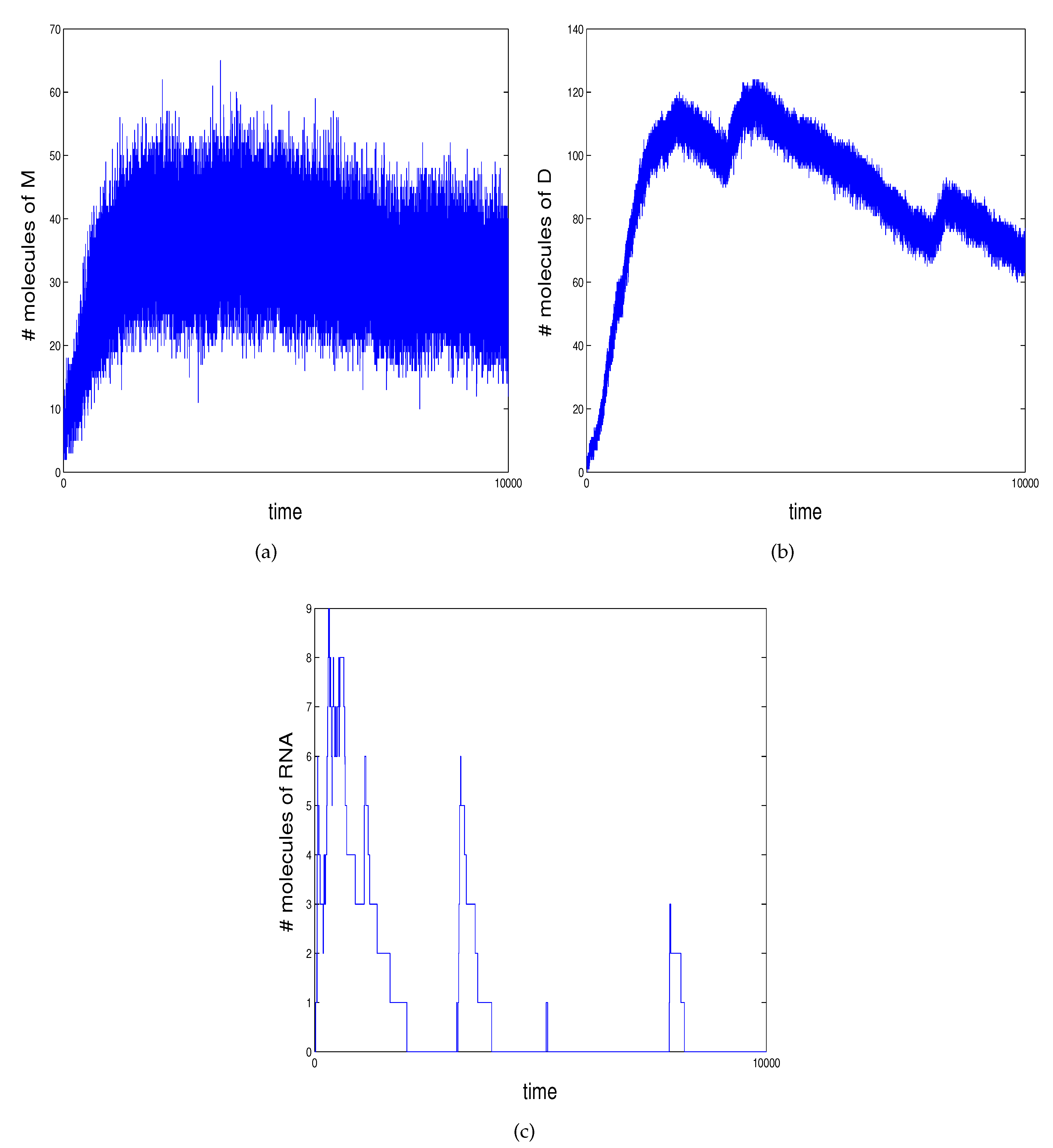
2.2. An Example of Genetic Regulation in Bacteriophage
- reaction 1 models transcription to mRNA occurring when D occupies ;
- reactions model mRNA translated into proteins and the decay of both;
- reaction 5 models protein dimerizing to the transcription factor D;
- reactions model activation of transcription of M when D binds at ;
- reactions 9 and 10 model prevention of the RNA polymerase from binding at the promoter region and repression of transcription when D binds at .
2.3. Demographic Noise in Bacteriophage Infections: The Backward Gene Regulation Approach
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mao, X. Stochastic stabilization and destabilization. Syst. Control Lett. 1994, 23, 279–290. [Google Scholar] [CrossRef]
- Mao, X. Stochastic self-stabilization. Stoch. Stoch. Rep. 1996, 57, 57–70. [Google Scholar] [CrossRef]
- Mao, X. Some contributions to stochastic asymptotic stability and boundedness via multiple Lyapunov functions. J. Math. Anal. Appl. 2001, 153, 325–340. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.J.S. An Introduction to Stochastic Processes with Applications to Biology, 2nd ed.; Chapman Hall/CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Allen, L.J.S. Stochastic Population and Epidemic Models. Persistence and Extinction; Mathematical Biosciences Lecture Series, Stochastics in Biological Systems; Springer: New York, NY, USA, 2015; Volume 1.3. [Google Scholar]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Gillespie, D.T. Approximate accelerated stochastic simulation of chemically reacting systems. J. Chem. Phys. 2001, 115, 1716. [Google Scholar] [CrossRef]
- Tian, T.; Burrage, K. Binomial leap methods for simulating stochastic chemical kinetics. J. Chem. Phys. 2004, 121, 10356–10364. [Google Scholar] [CrossRef]
- Cao, Y.; Li, H.; Petzold, L. Efficient formulation of the stochastic simulation algorithm for chemically reacting systems. J. Chem. Phys. 2004, 121, 4059–4067. [Google Scholar] [CrossRef]
- Cao, Y.; Gillespie, D.T.; Petzold, L.R. Avoiding negative populations in explicit Poisson tau-leaping. J. Chem. Phys. 2005, 123, 054104. [Google Scholar] [CrossRef]
- Cao, Y.; Gillespie, D.T.; Petzold, L.R. Efficient step size selection for the tau-leaping simulation method. J. Chem. Phys. 2006, 124, 044109. [Google Scholar] [CrossRef]
- Smith, H.L.; Trevino, R.T. Bacteriophage Infection Dynamics: Multiple Host Binding Sites. Math. Model. Nat. Phenom. 2009, 4, 109–134. [Google Scholar] [CrossRef]
- Mandal, P.S.; Allen, L.J.S.; Banerjee, M. Stochastic modeling of phytoplankton allelopathy. Appl. Math. Model. 2014, 38, 1583–1596. [Google Scholar] [CrossRef]
- Carletti, M.; Montani, M.; Meschini, V.; Radici, L.; Bianchi, M. Stochastic modeling of PTEN regulation in brain tumors. A model for glioblastoma multiforme. Math. Biosci. Eng. 2015, 12, 965–981. [Google Scholar] [PubMed]
- Beretta, E.; Kuang, Y. Modeling and analysis of a marine bacteriophage infection. Math. Biosci. 1998, 149, 57–76. [Google Scholar] [CrossRef]
- Burrage, K.; Tian, T. Effective simulation techniques for biological systems, in fluctuations and noise in biological, biophysical, and biomedical systems II. In Proceedings of the SPIE—The International Society for Optical Engineering: Fluctuations and Noise in Biological, Biophysical, and Biomedical Systems II, Maspalomas, Spain, 26–28 May 2004; SPIE: Bellingham, WA, USA, 2004; Volume 5467, pp. 311–325. [Google Scholar]
- Burrage, K.; Tian, T.; Burrage, P.M. A multi-scaled approach for simulating chemical reaction systems. Prog. Biophys. Mol. Biol. 2004, 85, 217–234. [Google Scholar] [CrossRef]
- Burrage, K.; Burrage, P.; Leier, A.; Marquez-Lago, T.A.; Marquez-Lago, T. A review of stochastic and delay simulation approaches in both time and space in computational cell biology. In Stochastic Processes, Multiscale Modeling, and Numerical Methods for Computational Cellular Biology; Holcman, D., Ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Goutsias, J. Quasiequilibrium approximation of fast reaction kinetics in stochastic biochemical sysmtems. J. Chem. Phys. 2005, 122, 184102. [Google Scholar] [CrossRef]
- Choudhuri, S. Gene Regulation and Molecular Toxicology. Toxicol. Mech. Methods 2004, 15, 1–23. [Google Scholar] [CrossRef]
- Haseltine, E.L.; Rawlings, J.B. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J. Chem. Phys. 2002, 117, 6959–6969. [Google Scholar] [CrossRef]
- Berry, H. Monte-Carlo simulations of enzyme reactions in two dimensions: Fractal kinetics and spatial segregation. Biophys. J. 2002, 83, 1891–1901. [Google Scholar] [CrossRef]
- Schnell, S.; Turner, T.E. Reaction kinetics in Intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophysica Mol. Biol. 2004, 85, 235–260. [Google Scholar] [CrossRef]
- Nicolau, D.V., Jr.; Burrage, K. Stochastic simulation of chemical reactions in spatially complex media. Comput. Math. Appl. 2008, 55, 1007–1018. [Google Scholar] [CrossRef]
- Arkin, A.; Ross, J.; McAdams, H.H. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics 1998, 149, 1633–1648. [Google Scholar] [PubMed]
- Elowitz, M.B.; Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Gonze, D.; Halloy, J.; Goldbeter, A. Robustness of circadian rythms with respect to molecular noise. Proc. Natl. Acad. Sci. USA 2002, 99, 673–678. [Google Scholar] [CrossRef]
- Gillespie, D.T. A rigorous derivation of the chemical master equation. Physica A 1992, 188, 404–425. [Google Scholar] [CrossRef]
- Ackers, G.K.; Johnson, A.D.; Shea, M.A. Quantitative model for gene regulation by λ phage repressor. Proc. Natl. Acad. Sci. USA 1982, 79, 1129–1133. [Google Scholar] [CrossRef]
- Hasty, J.; Pradines, J.; Dolnik, M.; Collins, J.J. Noise-based switches and amplifiers for gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 2075–2080. [Google Scholar] [CrossRef]
- Ptashne, M. A Genetic Switch: Phage λ Revisited, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004. [Google Scholar]
- Shea, M.A.; Ackers, G.K. The OR control system of bacteriophage Lambda: A physical-chemical model for gene regulation. J. Mol. Biol. 1985, 181, 211–230. [Google Scholar] [CrossRef]
- Barrio, M.; Burrage, K.; Leier, A.; Tian, T. Oscillatory Regulation of Hes1: Discrete stochastic delay modeling and simulation. PLoS Comput Biol. 2006, 2, e117. [Google Scholar] [CrossRef]
- Braumann, C.A. Environmental versus demographic stochasticity in population growth. In Workshop on Branching Processes and Their Applications; González, M.V., Puerto, I.M., Martínez, R., Molina, M., Mota, M., Ramos, A., Eds.; 37 Lecture Notes in Statistics—Proceedings; Springer: Berlin/Heidelberg, Germany, 2010; Volume 197. [Google Scholar]
- Vieira dos Santos, R. Discreteness inducing coexistence. Physica A 2013, 392, 5888–5897. [Google Scholar] [CrossRef][Green Version]
- Constable, G.W.A.; Rogers, T.; McKane, A.J.; Tarnita, C.E. Demographic noise can reverse the direction of deterministic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E4745–E4754. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, H.; Shnerb, N.M.; Kessler, D.A. Simulation of spatial systems with demographic noise. Phys. Rev. E 2018, 98, 022131. [Google Scholar] [CrossRef] [PubMed]
- Beretta, E.; Kuang, Y. Modeling and analysis of a marine bacteriophage infection with latency period. Nonlin. Anal. RWA 2001, 2, 35–74. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Aviramb, I.; Zaritsky, A. Bacterial debris—An ecological mechanism for coexistence of bacteria and their viruses. J. Theor. Biol. 2003, 224, 377–383. [Google Scholar] [CrossRef]
- Buckwar, E. The Θ-Maruyama scheme for stochastic functional differential equations with distributed memory term. Monte Carlo Methods Appl. 2004, 10, 235–244. [Google Scholar] [CrossRef]
- Gourley, S.A.; Kuang, Y. A stage structured predator-prey model and its dependence on maturation delay and death rate. J. Math. Biol. 2004, 49, 188–200. [Google Scholar] [CrossRef]
- Gourley, S.A.; Kuang, Y. A delay reaction-diffusion model of the spread of bacteriophage infection. SIAM J. Appl. Math. 2005, 65, 550–566. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Z.; Tang, J. A delayed marine bacteriophage infection model. Appl. Math. Lett. 2007, 20, 702–706. [Google Scholar] [CrossRef]
- Carletti, M. On the stability properties of a stochastic model for phage–bacteria interaction in open marine environment. Math. Biosci. 2002, 175, 117–131. [Google Scholar] [CrossRef]
- Carletti, M. Mean-square stability of a stochastic model for bacteriophage infection with time delays. Math. Biosci. 2007, 210, 395–414. [Google Scholar] [CrossRef]
- Leier, A.; Marquez-Lago, T.T.; Burrage, K. Generalized binomial Tau-leap method for biochemical kinetics incorporating both delay and intrinsic noise. J. Chem. Phys. 2008, 128, 205107. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Burrage, K.; Burrage, P.M.; Carletti, M. Stochastic delay differential equations for genetic regulatory networks. J. Comp. Appl. Math. 2007, 205, 696–707. [Google Scholar] [CrossRef]
- Beretta, E.; Carletti, M.; Solimano, F. On the effects of environmental fluctuations in a simple model of bacteria-bacteriophage interaction. Canad. Appl. Math. Quart. 2000, 8, 321–366. [Google Scholar]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carletti, M.; Banerjee, M. A Backward Technique for Demographic Noise in Biological Ordinary Differential Equation Models. Mathematics 2019, 7, 1204. https://doi.org/10.3390/math7121204
Carletti M, Banerjee M. A Backward Technique for Demographic Noise in Biological Ordinary Differential Equation Models. Mathematics. 2019; 7(12):1204. https://doi.org/10.3390/math7121204
Chicago/Turabian StyleCarletti, Margherita, and Malay Banerjee. 2019. "A Backward Technique for Demographic Noise in Biological Ordinary Differential Equation Models" Mathematics 7, no. 12: 1204. https://doi.org/10.3390/math7121204
APA StyleCarletti, M., & Banerjee, M. (2019). A Backward Technique for Demographic Noise in Biological Ordinary Differential Equation Models. Mathematics, 7(12), 1204. https://doi.org/10.3390/math7121204