

LiSENCE: A Hybrid Ligand and Sequence Encoder Network for Predicting CYP450 Inhibitors in Safe Multidrug Administration

 ,
,  , and
, and 
Abstract

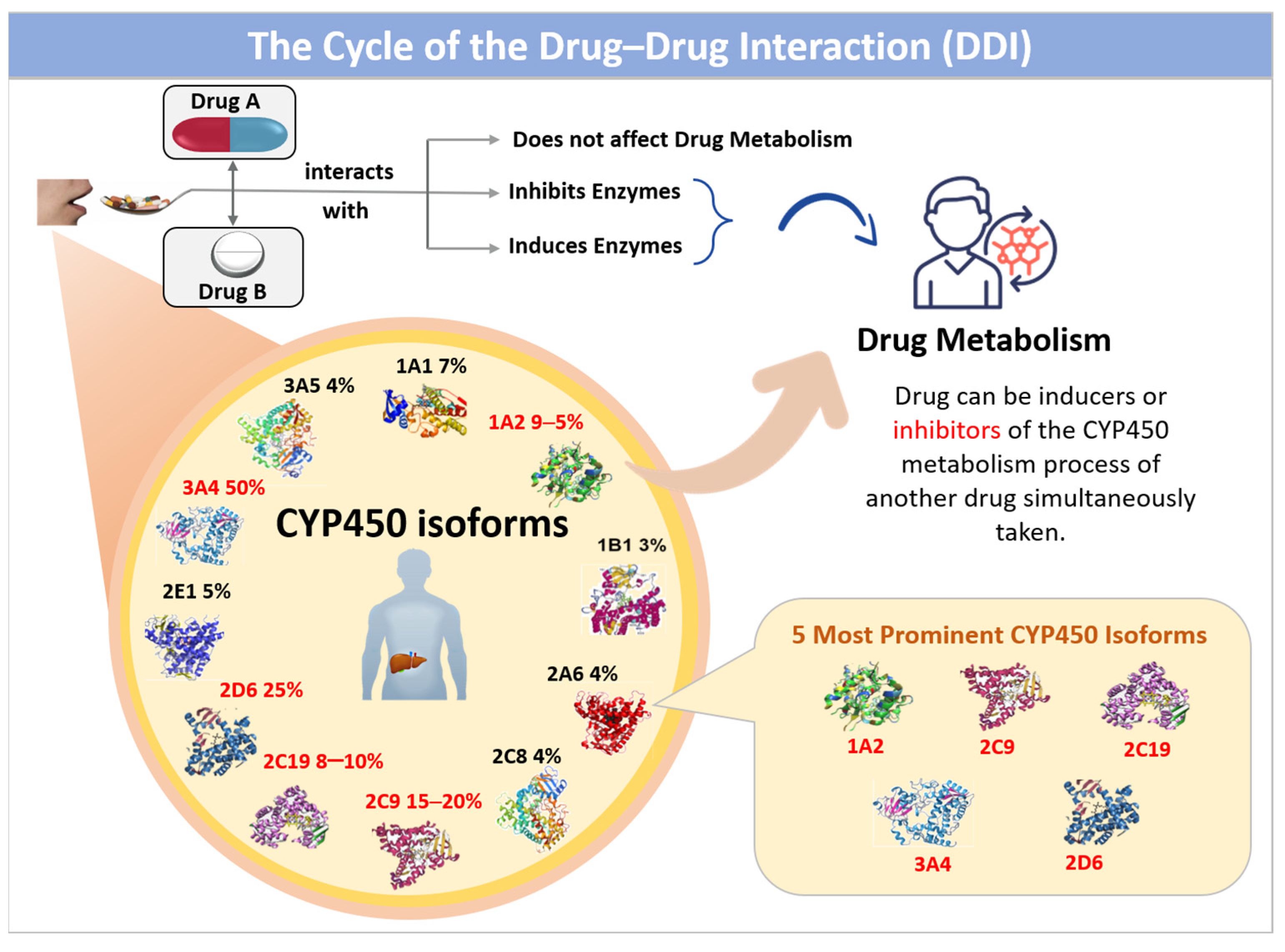
1. Introduction
- (1)
- A novel LiSENCE hybrid AI model is proposed to predict the CYP450 inhibitors by combining and assembling features from two pipelines: the Ligand Encoder Network (LEN) and the Sequence Encoder Network (SEN). Both pipelines incorporated various attention modules to enhance the feature extraction of ligands and protein target sequences.
- (2)
- To extract important node information from the ligand graph, a novel two-stage attention mechanism is proposed utilizing Joint-localized Attention (JoLA) and Self-Attention modules.
- (3)
- The innovative Attentive-GIN (AGIN) module is designed to enhance the extraction of the compound features.
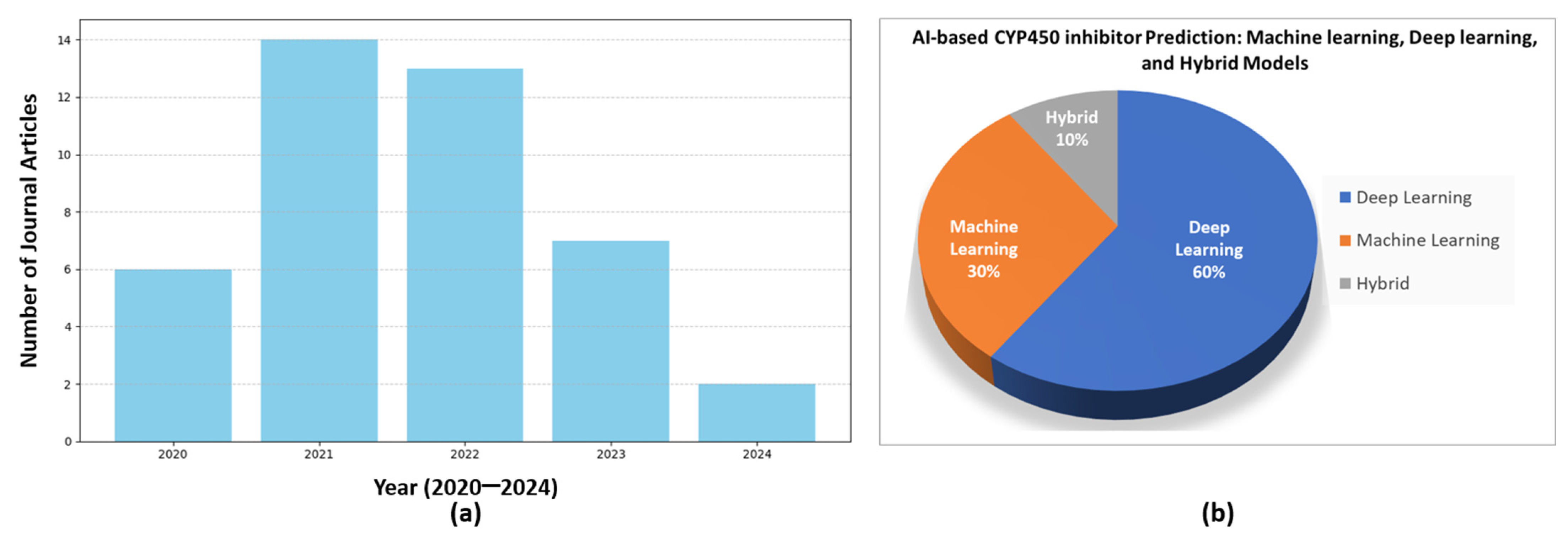
2. Related Work
3. Methods
3.1. Dataset
3.1.1. Drug/Ligand/Compound SMILEs String Dataset
PubChem Database
ChEMBL CYP450 BioAssay Data
3.1.2. CYP Target Protein Sequence Dataset
3.2. Preprocessing
3.3. The Proposed LiSENCE Framework for CYP450 Inhibitors Prediction
3.3.1. Ligand Encoder Network (LEN)
Joint-Localized Attention (JoLA)
Attentive Graph Isomorphism Network (AGIN)
Hybrid Pooling Layer
Multiple Convolution Layers
3.3.2. Sequence Encoder (SEN)
3.3.3. Classification Module
3.3.4. Explainable (XAI) Module
3.4. Evaluation Strategy
3.5. Training and Inference Settings
4. Experimental Results
4.1. Convergence of the Proposed AI Framework
4.2. LEN Evaluation Results
4.2.1. Examining JoLA
4.2.2. Examining AGIN
4.2.3. Examining LEN (End-to-End)
4.3. SEN Evaluation Results
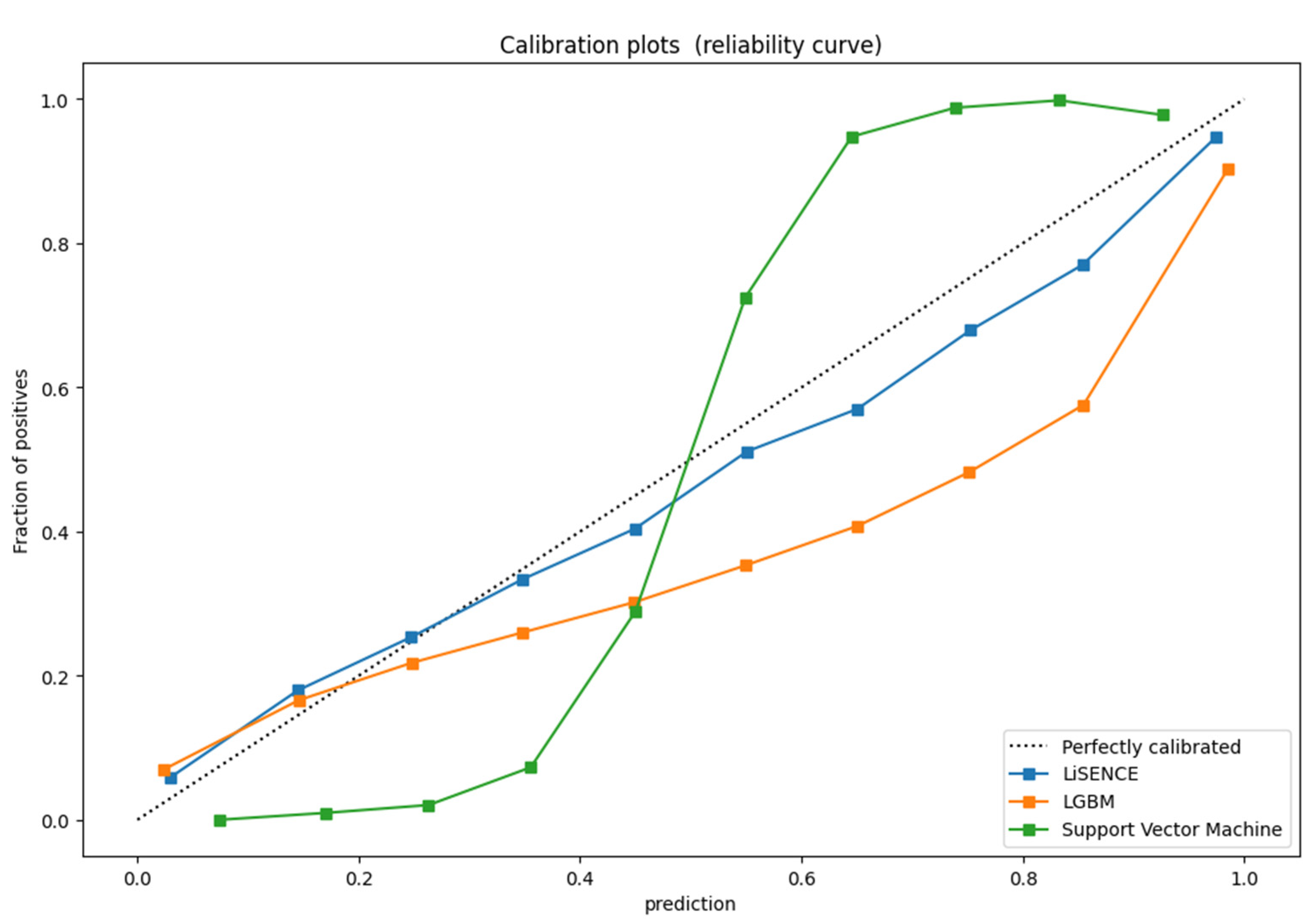
4.4. Evaluation Comparison Results of LiSENCE to Other State-of-the-Art Methods
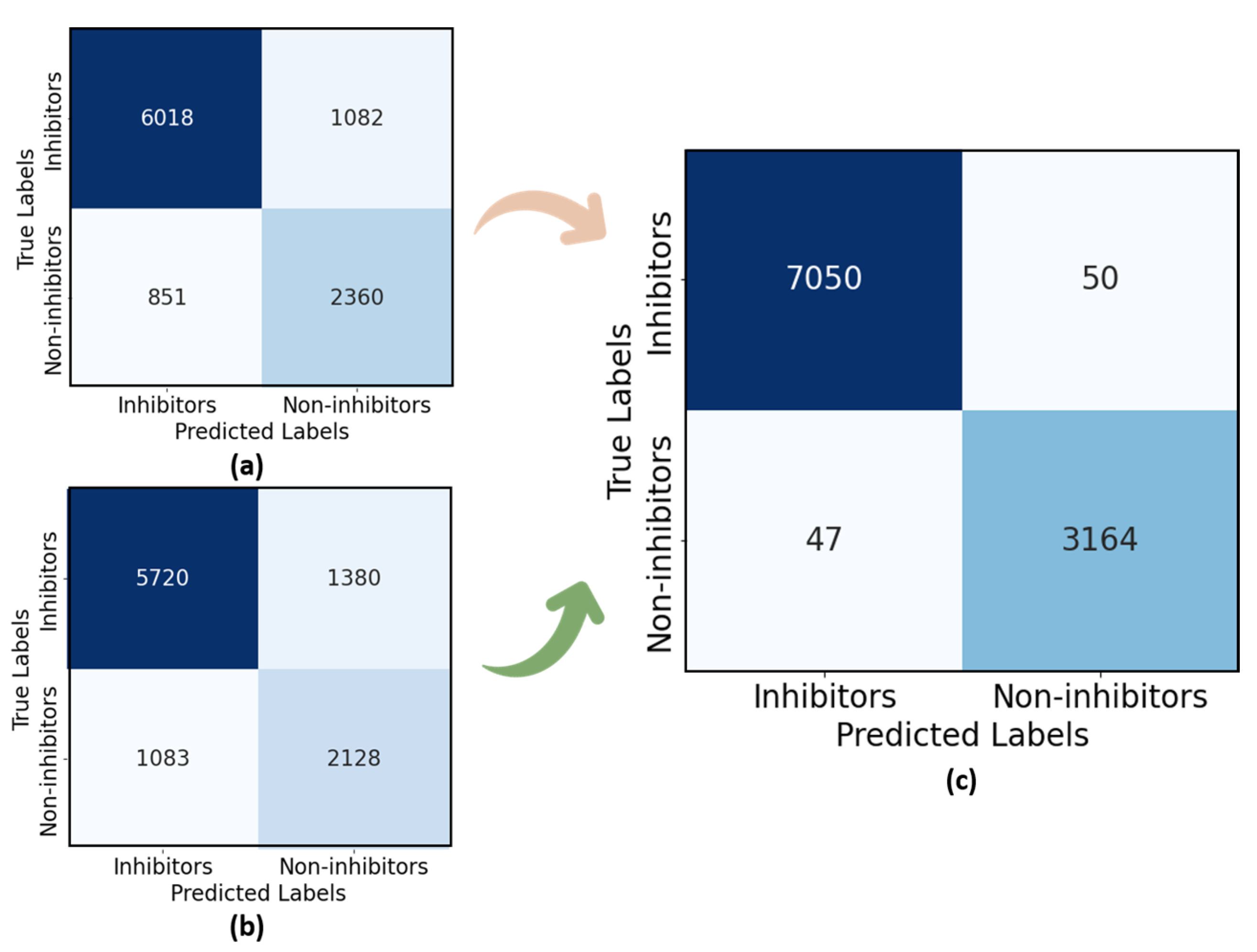
4.5. Visualizing Results with XAI Module and Re-Scoring LiSENCE
Re-Scoring LiSENCE After Explainability
4.6. Ablation Study Using ChEMBL External Dataset: Verification and Validation (V&V)
Statistical Testing of 10-Fold CV
4.7. Clinical Verification, Validation, and Application
5. Discussion
5.1. The Effectiveness of Various AI Configurations: Individual LEN Ablation Study
5.2. The Effectiveness of Various AI Configurations: Individual SEN Ablation Study
5.3. The Proposed LiSENCE Against Its Individual Sub-Models (LEN and SEN)
5.4. LiSENCE vs. the State-of-the-Art AI Models
5.5. Biological Deductions from XAI Module
5.6. Ablation Study: Biological Inference of Clinical Verification and Validation
6. Limitations
7. Future Works
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Artificial Intelligence |
| XAI | Explainable Artificial Intelligence |
| LEN | Ligand Encoder Network |
| DDI | Drug–Drug Interactions |
| SEN | Sequence Encoder Network |
| LiSENCE | Ligand and Sequence Encoder Networks for Predicting CYP450 Inhibitors |
| GIN | Graph Isomorphism Network |
| AGIN | Attentive Graph Isomorphism Network |
| LA | Local Attention |
| JoLA | Joint-localized Attention |
| ML | Machine Learning |
| CV | Cross-validation |
| DL | Deep Learning |
| BR | Before Re-scoring |
| AR | After Re-scoring |
| SMILES | Simplified Molecular Input Line Entry System |
| PDB | Protein Data Bank |
| CYP450 | Cytochrome P450 |
References
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, R. Computational models for predicting interactions with cytochrome p450 enzyme. Curr. Top. Med. Chem. 2006, 6, 1609–1618. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006, 8, E101–E111. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Liu, J.-P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Wang, B.; Yang, L.-P.; Liu, J.-P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 2010, 42, 268–354. [Google Scholar] [CrossRef]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Cassagnol, B.G.M. Biochemistry, Cytochrome P450. In StatPearls [Internet]; Statpearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Njimbouom, S.N.; Kim, J.-D. MuMCyp_Net: A multimodal neural network for the prediction of Cyp450. Expert Syst. Appl. 2024, 255, 124703. [Google Scholar] [CrossRef]
- Chang, J.; Fan, X.; Tian, B. DeepP450: Predicting Human P450 Activities of Small Molecules by Integrating Pretrained Protein Language Model and Molecular Representation. J. Chem. Inf. Model. 2024, 64, 3149–3160. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, L.; Zhang, P.; Guo, H.; Zhang, R.; Li, L.; Li, X. Prediction of Cytochrome P450 Inhibition Using a Deep Learning Approach and Substructure Pattern Recognition. J. Chem. Inf. Model. 2024, 64, 2528–2538. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Ai, D.; Cai, H.; Wei, J.; Zhao, D.; Chen, Y.; Wang, L. DEEPCYPs: A deep learning platform for enhanced cytochrome P450 activity prediction. Front. Pharmacol. 2023, 14, 1099093. [Google Scholar] [CrossRef]
- Li, L.; Lu, Z.; Liu, G.; Tang, Y.; Li, W. Machine Learning Models to Predict Cytochrome P450 2B6 Inhibitors and Substrates. Chem. Res. Toxicol. 2023, 36, 1332–1344. [Google Scholar] [CrossRef]
- Ouzounis, S.; Panagiotopoulos, V.; Bafiti, V.; Zoumpoulakis, P.; Cavouras DKalatzis, I.; Matsoukas, M.T.; Katsila, T. A Robust Machine Learning Framework Built Upon Molecular Representations Predicts CYP450 Inhibition: Toward Precision in Drug Repurposing. OMICS A J. Integr. Biol. 2023, 27, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Weiser, B.; Genzling, J.; Burai-Patrascu, M.; Rostaing, O.; Moitessier, N. Machine learning-augmented docking. 1. CYP inhibition prediction. Digit. Discov. 2023, 2, 1841–1849. [Google Scholar] [CrossRef]
- Qiu, M.; Liang, X.; Deng, S.; Li, Y.; Ke, Y.; Wang, P.; Mei, H. A unified GCNN model for predicting CYP450 inhibitors by using graph convolutional neural networks with attention mechanism. Comput. Biol. Med. 2022, 150, 106177. [Google Scholar] [CrossRef]
- Nguyen-Vo, T.-H.; Trinh, Q.H.; Nguyen, L.; Nguyen-Hoang, P.-U.; Nguyen, T.-N.; Nguyen, D.T.; Nguyen, B.P.; Le, L. iCYP-MFE: Identifying Human Cytochrome P450 Inhibitors Using Multitask Learning and Molecular Fingerprint-Embedded Encoding. J. Chem. Inf. Model. 2022, 62, 5059–5068. [Google Scholar] [CrossRef]
- Plonka, W.; Stork, C.; Šícho, M.; Kirchmair, J. CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes. Bioorganic Med. Chem. 2021, 46, 116388. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Brahma, R.; Shin, J.M.; Cho, K.H. Prediction of human cytochrome P450 inhibition using bio-selectivity induced deep neural network. Bull. Korean Chem. Soc. 2022, 43, 261–269. [Google Scholar] [CrossRef]
- Banerjee, P.; Dunkel, M.; Kemmler, E.; Preissner, R. SuperCYPsPred-a web server for the prediction of cytochrome activity. Nucleic Acids Res. 2021, 48, W580–W585. [Google Scholar] [CrossRef]
- Atwereboannah, A.A.; Wu, W.-P.; Nanor, E. Prediction of Drug Permeability to the Blood-Brain Barrier using Deep Learning. In Proceedings of the 4th International Conference on Biometric Engineering and Applications, ICBEA ’21, Taiyuan, China, 25–27 May 2021. [Google Scholar]
- Atwereboannah, A.A.; Wu, W.-P.; Ding, L.; Yussif, S.B.; Tenagyei, E.K. Protein-Ligand Binding Affinity Prediction Using Deep Learning. In Proceedings of the 18th International Computer Conference on Wavelet Active Media Technology and Information Processing (ICCWAMTIP), Chengdu, China, 17–19 December 2021; pp. 208–212. [Google Scholar]
- Agyemang, B.; Wu, W.-P.; Kpiebaareh, M.Y.; Lei, Z.; Nanor, E.; Chen, L. Multi-view self-attention for interpretable drug-target interaction prediction. J. Biomed. Inform. 2020, 110, 103547. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G.A. RDKit: Open-Source Cheminformatics. Release 2014.03.1; Zenodo: Munich, Germany, 2014. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Pawar, S.S.; Rohane, S.H. Review on Discovery Studio: An important Tool for Molecular Docking. Asian J. Res. Chem. 2021, 14, 1–3. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Bielka, H.; Sharon, N.; Australia, E.W. Nomenclature and symbolism for amino acids and peptides (Recommendations 1983). Pure Appl. Chem. 1984, 56, 595–624. [Google Scholar] [CrossRef]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. Int. J. Comput. Vis. 2020, 128, 336–359. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient-boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]
- Pisner, D.A.; Schnyer, D.M. Support vector machine. In Methods and Applications to Brain Disorders; Academic Press: New York, NY, USA, 2020; pp. 101–121. Available online: https://www.sciencedirect.com/science/article/pii/B9780128157398000067 (accessed on 18 April 2025).
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; pp. 278–282. [Google Scholar]
- Malik, S.; Harode, R.; Kunwar, A. XGBoost: A Deep Dive into Boosting. (Introduction Documentation); Simon Fraser University: Burnaby, BC, Canada, 2020; pp. 1–21. [Google Scholar]
- Yang, F.-J. An Implementation of Naive Bayes Classifier. In Proceedings of the 2018 International Conference on Computational Science and Computational Intelligence (CSCI), Las Vegas, NV, USA, 12–14 December 2018; pp. 301–306. [Google Scholar]
- Laaksonen, J.; Oja, E. Classification with learning k-nearest neighbors. In Proceedings of the International Conference on Neural Networks (ICNN’96), Washington, DC, USA, 3–6 June 1996. [Google Scholar]
- Igho-Osagie, E.; Puenpatom, A.; Williams, M.G.; Song, Y.; Yi, D.; Wang, J.; Berman, R.; Gu, M.; He, C. Prevalence of potential drug-drug interactions with ritonavir-containing COVID-19 therapy. J. Manag. Care Spéc. Pharm. 2023, 29, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Lazar, J.D.; Wilner, K.D. Drug Interactions with Fluconazole. Clin. Infect. Dis. 1990, 12, S327–S333. [Google Scholar] [CrossRef]
- Marcus, F.I. Drug interactions with amiodarone. Am. Heart J. 1983, 106, 924–930. [Google Scholar] [CrossRef]
- Kaeser, B.; Zandt, H.; Bour, F.; Zwanziger, E.; Schmitt, C.; Zhang, X. Drug-Drug Interaction Study of Ketoconazole and Ritonavir-Boosted Saquinavir. Antimicrob. Agents Chemother. 2009, 53, 609–614. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AI Category | Reference | Model /Platform | Dataset | Description | Limitation |
|---|---|---|---|---|---|
| Machine Learning | Li et al. [20], (2023) | RF, SVM, XGBoost | 500 compounds (CYP2B6 inhibitors vs. non-inhibitors) | Utilized molecular fingerprints, focusing on CYP2B6 | Single-isoform, single-feature type approach can be suboptimal for capturing isoform-specific patterns. |
| Ouzounis et al. [21], (2023) | Majority -voting ensemble (CloudScreen platform) | PubChem and ChEMBL data | Used multiple molecular representations for drug repurposing | Relies on protein docking to generate features, adding complexity and requiring known structures. Data for some isoforms (CYP2A6/2B6) is limited, which may affect those models’ reliability. | |
| Plonka et al. [25], (2021) | Random Forest models (web tool) | ChEMBL, PubChem, and a private ADME dataset | Random Forest classifiers for each of the 5 major CYP isoforms | Proprietary data inclusion means the model is not easily reproducible by others. | |
| Banerjee et al. [27], (2021) | Random Forest (web server) | 17,143 compounds from PubChem | Random Forest deployed as a web server to classify inhibitors CYP1A2, 2C9, 2C19, 2D6, 3A4 | Uses simple molecular fingerprints, offering limited interpretability. | |
| Deep Learning | Chen et al. [17] (2024) | Deep learning + Substructure Recognition | 85,715 compounds from PubChem BioAssay—high-quality curated set for 5 CYPs | Interpretable deep model highlighting key substructures | Requires a very large training set; performance on chemotypes outside the training distribution or on other CYP isoforms remains untested. |
| Ai et al. [19] (2023)—DEEPCYPs | Multitask FP-GNN (web server) | 65,467 compounds (after curation) from PubChem BioAssay | Fuses GCN with fingerprint features to predict CYP inhibition for 5 major CYP isoforms | Offers limited interpretability—it can highlight influential fragments, but the reasons for predictions are not transparent. | |
| Qiu et al. [23](2022) | GCNN + Attention | 17k compounds (PubChem inhibitor data for 5 CYPs, same dataset as iCYP-MFE) | Handles multiple CYP isoforms with end-to-end learning | Requires high-quality protein structure or sequence data for each isoform, limiting its application to well-characterized CYPs. | |
| Park et al. [26] (2022) | Bio-selectivity multitask DNN (stereochemical descriptors) | 150,000 compounds from PubChem (5 CYP isoforms) | A multitask deep neural network model for CYP inhibition that introduced 3D stereochemical descriptors to account for bio-selectivity differences among isoforms | Moreover, like other data-driven models, it does not explicitly model protein–ligand interactions, so mechanistic insight remains limited to correlations in the training data. | |
| Hybrid | Nguyen-Vo et al. [24] (2022)—iCYP-MFE | Multitask learning + Fingerprint Embedding | 17,143 compounds from PubChem (5 CYP isoforms)—curated and split for multitask learning | Graph-based learning with attention to relevant subgraphs | Uses a single type of input feature (chemical fingerprints) for all tasks, which can limit its ability to capture isoform-specific nuances. |
| Weiser et al. [22] (2023) | ML- Augmented Docking | Combined known inhibitor data for major CYPs | Incorporates selectivity profiles into model training | The workflow is computationally intensive (each query requires docking simulation). | |
| Kim et al. [18] (2023) | PubChem Resource | Millions of screening results (incl. CYP inhibition assays) | Learns cross-task CYP inhibition representations | Despite curation efforts, the translational relevance of high-throughput inhibition data (often single-concentration or in vitro) to clinical DDI risk can be limited. |
| Isoforms | Inhibitors | Non-Inhibitors | Ratio (Pos/Neg) | Total | ||||
|---|---|---|---|---|---|---|---|---|
| Training/ Validation | Testing | Training/ Validation | Testing | Training/ Validation | Testing | Training/ Validation | Testing | |
| 1A2 | 3885 | 158 | 6223 | 1120 | 0.14:1 | 0.62:1 | 10,108 | 1278 |
| 2C9 | 2808 | 7252 | 7089 | 27,502 | 0.26:1 | 0.40:1 | 9897 | 34,754 |
| 2C19 | 4599 | 8755 | 6101 | 22,758 | 0.38:1 | 0.75:1 | 10,700 | 31,513 |
| 2D6 | 1392 | 182 | 9698 | 1291 | 0.14:1 | 0.14:1 | 11,090 | 1473 |
| 3A4 | 3083 | 916 | 6668 | 2680 | 0.34:1 | 0.46:1 | 9751 | 3596 |
| Total | 15,767 | 17,263 | 35,779 | 55,351 | 0.31:1 | 0.44:1 | 51,546 | 72,614 |
| Isoforms | Inhibitors | Non-Inhibitors | Total |
|---|---|---|---|
| 1A2 | 578 | 1042 | 1620 |
| 2C9 | 1278 | 1434 | 2712 |
| 2C19 | 609 | 952 | 1561 |
| 2D6 | 1386 | 1740 | 3126 |
| 3A4 | 2572 | 2075 | 4647 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 30 | 65 | 28 | 45 | 18 | 25 | 26 |
| CI | (25.0, 35.0) | (59.5, 70.5) | (25.6, 30.4) | (40.1, 49.9) | (14.4, 21.6) | (21.4, 28.6) | (22.2, 29.8) |
| CYP2C9 | 34 | 67 | 30 | 47 | 19 | 27 | 28 |
| CI | (29.0, 39.0) | (61.5, 72.5) | (27.6, 32.4) | (42.1, 51.9) | (15.4, 22.6) | (23.4, 30.6) | (24.2, 31.8) |
| CYP2C19 | 36 | 69 | 31 | 50 | 20 | 29 | 30 |
| CI | (31.0, 41.0) | (63.5, 74.5) | (28.6, 33.4) | (45.1, 54.9) | (16.4, 23.6) | (25.4, 32.6) | (26.2, 33.8) |
| CYP2D6 | 39 | 71 | 32 | 52 | 21 | 30 | 31 |
| CI | (34.0, 44.0) | (65.5, 76.5) | (29.6, 34.4) | (47.1, 56.9) | (17.4, 24.6) | (26.4, 33.6) | (27.2, 34.8) |
| CYP3A4 | 44.5 | 76.5 | 33 | 55 | 25.5 | 32.5 | 34 |
| CI | (35.0, 45.0) | (71.0, 82.0) | (30.6, 35.4) | (50.1, 59.9) | (21.9,29.1) | (28.9, 36.1) | (30.2, 37.8) |
| Average | 35.8 | 69.7 | 30.8 | 49.8 | 20.7 | 28.7 | 29.8 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 35 | 68 | 35 | 53 | 22 | 25 | 30 |
| CI | (30.3, 39.7) | (63.3, 72.7) | (31.3, 38.7) | (48.3, 57.7) | (18.2, 25.8) | (20.7, 29.3) | (27.0, 33.0) |
| CYP2C9 | 37 | 70 | 37 | 55 | 24 | 27 | 31 |
| CI | (32.3, 41.7) | (65.3, 74.7) | (33.3, 40.7) | (50.3, 59.7) | (20.2, 27.8) | (22.7, 31.3) | (28.0, 34.0) |
| CYP2C19 | 40 | 72 | 39 | 57 | 26 | 30 | 33 |
| CI | (35.3, 44.7) | (67.3, 76.7) | (35.3, 42.7) | (52.3, 61.7) | (22.2, 29.8) | (25.7, 34.3) | (30.0, 36.0) |
| CYP2D6 | 42 | 75 | 41 | 60 | 27 | 32 | 34 |
| CI | (37.3, 46.7) | (70.3, 79.7) | (37.3, 44.7) | (55.3, 64.7) | (23.2, 30.8) | (27.7, 36.3) | (31.0, 37.0) |
| CYP3A4 | 44.5 | 77.5 | 42.5 | 62.5 | 30 | 33.5 | 36 |
| CI | (39.8, 49.2) | (72.8, 82.2) | (38.8, 46.2) | (57.8, 67.2) | (26.2, 33.8) | (29.2, 37.8) | (33.0, 39.0) |
| Average | 39.7 | 72.5 | 38.9 | 57.5 | 25.8 | 29.5 | 32.8 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 42 | 85 | 40 | 60 | 30 | 28 | 42 |
| CI | (38.9, 45.1) | (79.7, 90.3) | (35.7, 44.3) | (54.3, 65.7) | (24.0, 36.0) | (23.9, 32.1) | (38.3, 45.7) |
| CYP2C9 | 44 | 90 | 42 | 63 | 32 | 30 | 44 |
| CI | (40.9, 47.1) | (84.7, 95.3) | (37.7, 46.3) | (57.3, 68.7) | (26.0, 38.0) | (25.9, 34.1) | (40.3, 47.7) |
| CYP2C19 | 46 | 92 | 45 | 66 | 34 | 32 | 46 |
| CI | (42.9, 49.1) | (86.7, 97.3) | (40.7, 49.3) | (60.3, 71.7) | (28.0, 40.0) | (27.9, 36.1) | (42.3, 49.7) |
| CYP2D6 | 48 | 95 | 47 | 68 | 38 | 34 | 48 |
| CI | (44.9, 51.1) | (89.7, 98.3) | (42.7, 51.3) | (62.3, 73.7) | (32.0, 44.0) | (29.9, 38.1) | (44.3, 51.7) |
| CYP3A4 | 47.5 | 95.5 | 48.5 | 72 | 42 | 36.5 | 49.5 |
| CI | (44.4, 50.6) | (90.2, 97.8) | (44.2, 52.8) | (66.3, 77.7) | (36.0, 48.0) | (32.4, 40.6) | (45.8, 53.2) |
| Average | 45.5 | 91.5 | 44.5 | 65.8 | 35.2 | 32.1 | 45.9 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 34 | 85 | 50 | 65 | 20 | 34 | 74 |
| CI | (29.7, 38.3) | (80.5, 89.5) | (47.2, 52.8) | (60.8, 69.2) | (17.2, 22.8) | (30.3, 37.7) | (68.7, 79.3) |
| CYP2C9 | 36 | 88 | 52 | 68 | 21 | 36 | 76 |
| CI | (31.7, 40.3) | (83.5, 92.5) | (49.2, 54.8) | (63.8, 72.2) | (18.2, 23.8) | (32.3, 39.7) | (70.7, 81.3) |
| CYP2C19 | 38 | 90 | 54 | 70 | 22 | 38 | 78 |
| CI | (33.7, 42.3) | (85.5, 94.5) | (51.2, 56.8) | (65.8, 74.2) | (19.2, 24.8) | (34.3, 41.7) | (72.7, 83.3) |
| CYP2D6 | 40 | 92 | 56 | 72 | 24 | 40 | 82 |
| CI | (35.7, 44.3) | (87.5, 96.5) | (53.2, 58.8) | (67.8, 76.2) | (21.2, 26.8) | (36.3, 43.7) | (76.7, 87.3) |
| CYP3A4 | 43 | 94.5 | 52.5 | 73.5 | 25.5 | 41.5 | 84.5 |
| CI | (38.7, 47.3) | (90.0, 99.0) | (49.7, 55.3) | (69.3, 77.7) | (22.7, 28.3) | (37.8, 45.2) | (79.2, 89.8) |
| Average | 38.2 | 89.9 | 52.9 | 69.7 | 22.5 | 37.9 | 78.9 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 82 | 89 | 70 | 85 | 68.1 | 74 | 85 |
| CI | (79.2, 84.8) | (87.4, 90.6) | (66.6, 73.4) | (82.0, 88.0) | (66.3, 69.9) | (67.1, 80.9) | (80.8, 89.2) |
| CYP2C9 | 84 | 92 | 74 | 88 | 71.5 | 78 | 87 |
| CI | (81.2, 86.8) | (90.4, 93.6) | (70.6, 77.4) | (85.0, 91.0) | (69.7, 73.3) | (71.1, 84.9) | (82.8, 91.2) |
| CYP2C19 | 85 | 91 | 72 | 90 | 70.3 | 64 | 80 |
| CI | (82.2, 87.8) | (89.4, 92.6) | (68.6, 75.4) | (87.0, 93.0) | (68.5, 72.1) | (57.1, 70.9) | (75.8, 84.2) |
| CYP2D6 | 80 | 90 | 76 | 84 | 69.7 | 75 | 89 |
| CI | (77.2, 82.8) | (88.4, 91.6) | (72.6, 79.4) | (81.0, 87.0) | (67.9, 71.5) | (68.1, 81.9) | (84.8, 93.2 |
| CYP3A4 | 85.5 | 92 | 76.5 | 86.5 | 71.5 | 76.5 | 85.5 |
| CI | (82.7, 88.3) | (90.4, 93.6) | 73.1,79.9) | (83.5, 89.5) | (69.7, 73.3) | (69.6, 83.4) | (81.3, 89.7) |
| Average | 70.1 | 95.8 | 81.7 | 92.2 | 70.3 | 76.7 | 86.9 |
| Isoforms | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 65 | 88 | 74 | 88 | 60 | 70 | 77 |
| CI | (62.4, 67.6) | (84.1, 91.9) | (69.9, 78.1) | (84.2, 91.8) | (54.3, 65.7) | (67.0, 73.0) | (73.9, 80.1) |
| CYP2C9 | 67 | 89 | 76 | 90 | 63 | 72 | 79 |
| CI | (64.4, 69.6) | (85.1, 92.9) | (71.9, 80.1) | (86.2, 93.8) | (57.3, 68.7) | (69.0, 75.0) | (75.9, 82.1) |
| CYP2C19 | 66 | 91 | 78 | 92 | 66 | 74 | 81 |
| CI | (63.4, 68.6) | (87.1, 94.9) | (73.9, 82.1) | (88.2, 95.8) | (60.3, 71.7) | (71.0, 77.0) | (77.9, 84.1) |
| CYP2D6 | 64 | 92 | 80 | 93 | 68 | 75 | 83 |
| CI | (61.4, 66.6) | (88.1, 95.9) | (75.9, 84.1) | (89.2, 96.8) | (62.3, 73.7) | (72.0, 78.0) | (79.9, 86.1) |
| CYP3A4 | 69.5 | 96 | 82.5 | 96 | 72 | 76 | 82.5 |
| CI | (66.9, 72.1) | (92.1, 99.9) | (78.4, 86.6 | 92.2, 99.8) | (66.3, 77.7) | (73.0, 79.0) | (79.4, 85.6) |
| Average | 66.3 | 91.2 | 78.1 | 91.8 | 65.8 | 73.4 | 80.5 |
| CYP | Reference, (Year) | Method | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | Njimbouom et al. [15] (2024) | MumCypNet | 82 | 90 | 82 | 85 | 64 | 82 | 82 |
| Ai et al. [19], (2023) | DEEPCYPs | 84 | 93 | 82 | 88 | 70 | 82 | 91 | |
| Qiu et al. [23], (2022) | GCNN | 84 | 92 | 82 | 86 | 68 | 84 | 92 | |
| Ngyuyen et al. [24] (2021) | iCYP-MFE | 81 | 91 | 81 | 81 | 62 | 83 | 83 | |
| Ours, (2025) | LiSENCE | 87 | 95 | 85 | 92 | 68 | 86 | 81 | |
| 2C9 | Njimbouom et al. [15] (2024) | MumCypNet | 80 | 89 | 69 | 91 | 59 | 78 | 88 |
| Ai et al. [19] (2023) | DEEPCYPs | 84 | 91 | 72 | 93 | 66 | 76 | 85 | |
| Qiu et al. [23] (2022) | GCNN | 76 | 89 | 63 | 92 | 58 | 70 | 79 | |
| Ngyuyen et al. [24] (2021) | iCYP-MFE | 86 | 92 | 72 | 91 | 67 | 80 | 90 | |
| Ours, (2025) | LiSENCE | 87 | 93 | 79 | 80 | 66 | 78 | 85 | |
| 2C19 | Njimbouom et al. [15] (2024) | MumCypNet | 80 | 89 | 69 | 91 | 59 | 78 | 88 |
| Ai et al. [19] (2023) | DEEPCYPs | 78 | 86 | 71 | 84 | 57 | 76 | 81 | |
| Qiu et al. [23] (2022) | GCNN | 81 | 88 | 77 | 82 | 60 | 76 | 89 | |
| Ngyuyen et al. [24] (2021) | iCYP-MFE | 90 | 90 | 67 | 89 | 68 | 62 | 80 | |
| Ours, (2025) | LiSENCE | 92 | 91 | 80 | 92 | 67 | 77 | 90 | |
| 2D6 | Njimbouom et al. [15] (2024) | MumCypNet | 78 | 86 | 71 | 85 | 57 | 76 | 81 |
| Ai et al. [19] (2023) | DEEPCYPs | 91 | 88 | 64 | 91 | 60 | 65 | 78 | |
| Qiu et al. [23] (2022) | GCNN | 85 | 92 | 77 | 85 | 66 | 76 | 87 | |
| Ngyuyen et al. [24] (2021) | iCYP-MFE | 82 | 91 | 78 | 84 | 64 | 77 | 86 | |
| Ours, (2025) | LiSENCE | 92 | 95 | 79 | 81 | 65 | 76 | 87 | |
| 3A4 | Njimbouom et al. [15] (2024) | MumCypNet | 87 | 93 | 74 | 86 | 64 | 79 | 89 |
| Ai et al. [19] (2023) | DEEPCYPs | 82 | 90 | 82 | 85 | 64 | 82 | 82 | |
| Qiu et al. [23] (2022) | GCNN | 84 | 93 | 82 | 88 | 70 | 82 | 91 | |
| Ngyuyen et al. [24] (2021) | iCYP-MFE | 84 | 92 | 82 | 86 | 68 | 84 | 92 | |
| Ours, (2025) | LiSENCE | 88 | 95 | 83 | 87 | 68 | 85 | 93 | |
| Average Results of LiSENCE for all 5 isoforms | 89.2 | 97 | 92.2 | 97.3 | 87.8 | 83.3 | 93.8 | ||
| Isoforms | Re-Scoring | Acc | AUC | Sen | Spe | MCC | F1-Score | Pre |
|---|---|---|---|---|---|---|---|---|
| CYP1A2 | before | 87 | 95 | 85 | 92 | 68 | 86 | 81 |
| (84.0, 90.0) | (92.9, 97.1) | (80.6, 89.4) | (85.7, 98.3) | (65.3, 70.7) | (80.0, 92.0) | (75.5, 86.5) | ||
| after | 88 | 97 | 87 | 93 | 70 | 87 | 83 | |
| (85.0, 91.0) | (94.9, 99.1) | (82.6, 91.4) | (86.7, 99.3) | (67.3, 72.7) | (81.0, 93.0) | (77.5, 88.5) | ||
| CYP2C9 | before | 87 | 93 | 79 | 80 | 66 | 78 | 85 |
| (84.0, 90.0) | (90.9, 95.1) | (74.6, 83.4) | (73.7, 86.3) | (63.3, 68.7) | (72.0, 84.0) | (79.5, 90.5) | ||
| after | 88 | 92 | 80 | 84 | 67 | 74 | 82 | |
| (85.0, 91.0) | (89.9, 94.1) | (75.6, 84.4) | (77.7, 90.3) | (64.3, 69.7) | (68.0, 80.0) | (76.5, 87.5 | ||
| CYP2C19 | before | 92 | 91 | 80 | 92 | 67 | 77 | 90 |
| (63.4, 68.6) | (87.1, 94.9) | (73.9, 82.1) | (88.2, 95.8) | (60.3, 71.7) | (71.0, 77.0) | (77.9, 84.1) | ||
| after | 92 | 94 | 82 | 93 | 69 | 79 | 93 | |
| (89.0, 95.0) | (91.9, 96.1) | (77.6, 86.4) | (86.7, 99.3) | (66.3, 71.7) | (73.0, 85.0) | (87.5, 98.5) | ||
| CYP2D6 | before | 92 | 95 | 88 | 81 | 65 | 76 | 87 |
| (89.0, 95.0) | (92.9, 97.1) | (83.6, 92.4) | (74.7, 87.3) | (62.3, 67.7) | (70.0, 82.0) | (81.5, 92.5) | ||
| after | 93 | 93 | 89 | 83 | 63 | 78 | 88 | |
| (90.0, 96.0) | (90.9, 95.1) | (84.6, 93.4) | (76.7, 89.3) | (60.3, 65.7) | (72.0, 84.0) | (82.5, 93.5) | ||
| CYP3A4 | before | 88 | 95 | 83 | 87 | 68 | 85 | 93 |
| (85.0, 91.0) | (78.6, 87.4) | (78.6, 87.4) | (80.7, 93.3) | (65.3, 70.7) | (79.0, 91.0 | (87.5, 98.5) | ||
| after | 88 | 94 | 85 | 89 | 70 | 86 | 91 | |
| (85.0, 91.0) | (91.9, 96.1) | (80.6, 89.4) | (82.7, 95.3) | (67.3, 72.7) | (80.0, 92.0) | (85.5, 96.5) |
| Drugs | CYP450 Inhibits |
|---|---|
| Fluconazole | 2C9 and 3A4 |
| Clarithromycin | 3A4 |
| Cimetidine | 1A2, 2C19, 2D6, 3A4 |
| Ritonavir | 3A4 and 2D6 |
| Fluvoxamine | 1A2, 2C19 |
| Ketoconazole | 3A4 |
| Valproic Acid | 2C9, 2C19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atwereboannah, A.A.; Wu, W.-P.; Yussif, S.B.; Abdullah, M.A.; Tenagyei, E.K.; Ukuoma, C.C.; Gu, Y.H.; Al-antari, M.A. LiSENCE: A Hybrid Ligand and Sequence Encoder Network for Predicting CYP450 Inhibitors in Safe Multidrug Administration. Mathematics 2025, 13, 1376. https://doi.org/10.3390/math13091376
Atwereboannah AA, Wu W-P, Yussif SB, Abdullah MA, Tenagyei EK, Ukuoma CC, Gu YH, Al-antari MA. LiSENCE: A Hybrid Ligand and Sequence Encoder Network for Predicting CYP450 Inhibitors in Safe Multidrug Administration. Mathematics. 2025; 13(9):1376. https://doi.org/10.3390/math13091376
Chicago/Turabian StyleAtwereboannah, Abena Achiaa, Wei-Ping Wu, Sophyani B. Yussif, Muhammed Amin Abdullah, Edwin K. Tenagyei, Chiagoziem C. Ukuoma, Yeong Hyeon Gu, and Mugahed A. Al-antari. 2025. "LiSENCE: A Hybrid Ligand and Sequence Encoder Network for Predicting CYP450 Inhibitors in Safe Multidrug Administration" Mathematics 13, no. 9: 1376. https://doi.org/10.3390/math13091376
APA StyleAtwereboannah, A. A., Wu, W.-P., Yussif, S. B., Abdullah, M. A., Tenagyei, E. K., Ukuoma, C. C., Gu, Y. H., & Al-antari, M. A. (2025). LiSENCE: A Hybrid Ligand and Sequence Encoder Network for Predicting CYP450 Inhibitors in Safe Multidrug Administration. Mathematics, 13(9), 1376. https://doi.org/10.3390/math13091376