1. Introduction
Biological problems of reconstructing genes, genomes, genome syntenies, cell types and their annotations take up a central place in modern genomics. These biological problems are addressed in hundreds of publications, of which we would like to highlight reviews [
1,
2]. Well-known algorithms for such reconstruction are of a heuristic nature; most notably, they are not accompanied with evidential estimations of the algorithm’s accuracy and error in standard form; such mathematical questions are not even usually addressed in these publications. Among the algorithms of genome reconstruction, we highlight [
3] a convenient way of visualizing reconstructed genomes when working with databases [
4]. In regard to the biological problem of reconstructing genome syntenies, we highlight the review [
5]; and among the algorithms of its solution [
6,
7]. On reconstructing cell types, we highlight the work presented in [
8,
9]; in the latter, a network class is introduced with node marks that are convenient for describing cell types and their evolution.
From these biological reconstruction problems, and especially from the problem of reconstructing genome syntenies, a mathematical reconstruction problem arose along the network of graphs of a special type, called genomic structures. Recall that a genomic structure (hereafter referred to simply as a structure) is any directed graph whose components are paths and cycles (including loops, but not isolated nodes), and whose edges are assigned with names, for example, natural numbers. In other words, the degree of each node of the graph is either 1 or 2. Such a representation of a biological genome as a set of linear and circular chromosomes has been studied in biology for about thirty years. The formulation of the corresponding mathematical problem is based on a fixed set of SCJ or DCJ operations, each of which is assigned a cost, a strictly positive rational number.
Recall that
SCJ (Single-Cut-or-Join) operations are the following: a merge (join) of two free extremities of edges at one node of the graph and a
cut (the inverse of a join, cutting a node of degree 2); in the case of unequal sets of edge names of structures, two more operations are added here:
insertion of an isolated edge and its
deletion; see [
10]. The set of edge names is called the content of a structure.
Recall that DCJ (Double-Cut-and-Join) operations are as follows:
- (1)
Double inter-merging. Cut two nodes in the structure and merge (join) the four resulting free extremities.
- (2)
Sesquialteral inter-merging. Cut a node and merge (join) one resulting in free extremity with any free extremity in the structure.
- (3–4)
Single inter-mergings:
cut and
join (mutually inverse operations): undo a merge of extremities at one node, or join two free extremities together; see [
11].
As above, if structures a and b have unequal sets of edge names (say: unequal contents), two more operations are added here: insertion of a connected fragment of edges belonging to b but not to a into a component of a (or as its separate component), and deletion of such a fragment of edges belonging to a but not to b from a.
Thus, equal gene content of two genomic structures means that they have the same number of genes and a one-to-one orthology correspondence is defined between them. In other words, the names of edges in the structures are unique, and orthologous genes have identical names, i.e., names are not repeated in the structure, and the two sets of structure names coincide. In what follows, unless otherwise specified, we assume that names in a structure are unique. Recall that each operation is assigned with its cost, a strictly positive rational number.
In that way, we come to the definitions. The
transformation problem is as follows: for given structures
a and
b, find a minimal (in the total cost of operations) chain of
DCJ operations transforming
a into
b. For
SCJ operations this problem is trivial, but for
DCJ operations several dozens of works have been published; we mention below several main contributions. The
reconstruction problem for structures defined at leaves of a given network consists of arranging structures at non-leaf nodes of the tree so as to minimize the sum, over all its edges, of the
DCJ (or
SCJ) distance between the structures assigned to the endpoints of an edge. Here, the
DCJ distance is assumed to be the total cost of the minimal (in the total cost of operations)
DCJ transformation of the structure closer to the root to the structure closer to the leaves of the tree. The
SCJ distance is defined similarly. Actually, the transformation problem is a particular case of the reconstruction problem when a network consists of a single edge. Let us highlight the work presented in [
12], which introduces and studies the intermediary structure distance between
SCJ and
DCJ.
Despite a number of mathematical results, both mathematical problems remain completely nontrivial and far from being finally solved (especially the second one), regardless of their specific applications. In the bioinformatics context, an edge of a structure with its name is called a gene, and a component of the structure is called a chromosome. The structure itself is sometimes referred to as the genome. Describing a biological genome as a structure, of course, neglects most of its actual features but, nevertheless, it is useful to study the properties of the biological genome. One of the reasons for this usefulness is that structures allow efficient algorithms that are guaranteed to cope with hundreds of genes simultaneously. In essence, a structure describes the gene synteny, and algorithms allow us, among other things, to reconstruct the gene synteny and to identify ancient and stable syntenies and their evolutions.
In this publication, authors study the second problem—the reconstruction problem—specifically as a mathematical one, not touching on its multiple applications. The mathematical hardness of this problem is described through the fact that for the
DCJ distance, the reconstruction problem is NP-hard even for a tree consisting of a root and three leaves with equal gene content [
13]. For the
SCJ distance, the reconstruction problem seems simpler, although the authors suppose that it is also NP-hard for arbitrary operation costs and even for equal gene content.
In any case, its mathematical solution with a low-polynomial algorithm looks like an extremely interesting and tough challenge in the field of discrete optimization.
To conclude the Introduction, we mention a few papers that have affected our interest in this biological subject, first on transformation and then on reconstruction problems.
Among the pioneers who set and studied these and related problems, we should point out the works of D. Sankoff, P.A. Pevzner, and their students, referring to surveys [
14,
15,
16].
In the work presented in [
17], genomic rearrangements in ancestors of twenty modern mitochondrial genomes were studied. In the work presented in [
18], genomes consisting of a single chromosome and a reversal operation were considered, and the shortest sequence of reversals reducing an initial sequence of integers to a strictly increasing sequence of positive numbers was constructed. The resulting algorithm has degree-four polynomial runtime. In the work presented in [
19] the problem of determining the distance between two genomes was considered in which the chromosome is a sequence of nonzero integers. The operations of fusion, fission, reversal, and translocation were allowed; the corresponding algorithm with degree-four polynomial complexity was obtained. In the work presented in [
20], an important original construction of a breakpoint graph was proposed, in which we generalized to the case of unequal gene content [
21]. In the work presented in [
19], a reduction of the genome transformation problem to the problem of reducing a breakpoint graph to a final form was proposed, which we later generalized to the case of unequal gene content [
21].
In the work presented in [
11], a linear-time algorithm was proposed for solving the transformation problem with equal gene content of structures and equal operation costs (i.e., in their absence). In the work presented in [
22,
23], linear-time algorithms are described for solving this problem under unequal gene content and equal operation costs. In the work presented in [
24,
25,
26] linear-time algorithms are described for solving this problem with unequal gene content and partially unequal operation costs: the costs of the first four
DCJ operations are equal; the costs of insertion and deletion operations are also equal but possibly different from the cost of the first four operations. In the work presented in [
21,
27] a linear-time algorithm is described for solving this problem with unequal gene content and unequal operation costs: the costs of the first four
DCJ operations are equal (denote them by
c), the costs
wi and
wd of insertion and deletion operations may be unequal to each other and unequal to
c. If
c ≤ min(
wi,
wd) < max(
wi,
wd), then our algorithm may admit an additive error
k ≤ 2
c. If min(
wi,
wd) <
c < max(
wi,
wd), then we have
k ≤ max(
wi,
wd) −
c (if
wd +
wi ≤ 2
c),
k ≤ 4·max(
wi,
wd) + 2·min(
wi,
wd) − 6
c (if
wd +
wi > 2
c and max(
wi,
wd) ≤ 2
c) and
k ≤ 6·max(
wi,
wd) + 2·min(
wi,
wd) − 9
c (if max(
wi,
wd) > 2
c).
A separate difficult question is whether topological properties of structures
a and
b can be preserved in intermediate structures of the shortest (or close to it) transformation of
a into
b? In the case of equal gene content of
a and
b, the following partial results have been obtained [
28] (Section 6):
- (1)
Let structures a and b have equal content, and let each of them consist of a single cycle. There exists a quadratic-time algorithm which outputs a shortest transformation of a into b such that every intermediate structure in it consists of at most two cycles; if the second cycle appears at some step, then at the next step there remains a single cycle again.
- (2)
Let structures a and b have equal content, and let each of them consist of a single path. There exists a quadratic-time algorithm which outputs the shortest transformation of a into b such that any intermediate structure in it consists of at most one path and one cycle; if a cycle appears, then at the next step there remains a single path again.
- (3)
Let structures a and b have equal content, and let each of them consist of paths. There exists a quadratic-time algorithm which outputs the shortest transformation of a into b such that any intermediate structure in it consists of at most one cycle; if a cycle appears, then at the next step there remain only paths again.
Let us now touch upon the case where different edges of a structure can have equal names, i.e., there are paralogs in the structure. Then, the transformation problem is NP-hard for
SCJ operations [
29,
30] and for
DCJ operations [
31,
32]. A reduction of this problem to an integer linear programming problem is known, e.g., in the work presented in [
33]. Its direct solution (i.e., not by reducing this problem to another one) by an algorithm of polynomial complexity assumes constraints imposed on the initial structures and/or on the list of operations over structure. For example, in the work presented in [
34], paralogs are not allowed in structure
a but are allowed in structure
b, sets of names in
a and
b are equal (so-called “quasi-equal content”), while only cut and join operations are allowed and, additionally, an additional operation of edge duplication: inserting a new edge next to a given edge, with the same name and orientation (“linear duplication”), or adding an edge with the same name in the form of a loop as a separate component of the structure (“cyclic duplication”). A reminder here: the edges of the structure that have identical names are called
paralogs; the results of this publication relate to structures without paralogs, even though those are sometimes mentioned in the description of other authors’ publications. Item [
35] describes approximated algorithms for calculating various distances between paralog structures, considering not only their genetic composition, but also the distance between neighboring genes. Algorithm error scores proved to be proportional to the maximum number of gene paralogs in a structure.
We considered another constraint on
a and
b with quasi-equal content: as above,
a has no paralogs, and
b has at most two paralogs of each edge; however, all
DCJ operations over the structure are allowed together with the duplication operation, and costs of the operations are arbitrary [
28] (Section 1). This problem is already NP-hard [
28]. For this problem, the authors have obtained an algorithm that constructs a transformation of
a into
b with multiplicative error of at most 13/9 +
ε, where
ε is any strictly positive number; its runtime is of the order of
, where
n is the size of the initial pair of graphs
a and
b [
28].
Item [
36] describes the algorithm for solving the reconstruction problem for structures with an equal genetic composition on a tree that consists of a root and three leaves; at that,
DCJ operations are used, and a certain evaluation of the algorithm’s accuracy is received. In the work presented in [
37] (Section 3.6), a reduction of the reconstruction problem to a sequence of SAT problems is described for a star, i.e., a tree consisting of a root and arbitrarily many leaves. There, we assumed structures consisting of circular chromosomes, equal gene content, and, of course, using the double inter-merging operation only. However, for special cases, efficient algorithms are known. For example, in the work presented in [
10], an exact quadratic-time algorithm for structure reconstruction is described for equal
SCJ operation costs and equal content of structures. In the work presented in [
13], an algorithm is proposed for solving this problem for a star under equal
SCJ operation costs and equal content of structures. In [
34] this algorithm, also with equal costs, was generalized to the case of quasi-equal content (no paralogs at the root) and the operations of cut, join, and duplication. The runtimes of these algorithms are near quadratic. In the work presented in [
28] (Section 7), the authors described a (quadratic-time) algorithm for solving the reconstruction problem for a star with equal content of structures and arbitrary costs, or with unequal content but with no paralogs and no loops in the structures. This algorithm admits the requirement that all intermediate structures are cyclic if all given and sought-for structures consist of cycles (cyclic structures). There, the authors have also generalized this algorithm to the case of paralogs in leaf structures, obtaining the following result. Let us provide a star and structures at its leaves with quasi-equal content (i.e., paralogs are allowed). A structure with the same content and without paralogs is searched for at the root, and operations over the structures are cut, join, and duplication (linear and cyclic). Then, by a quadratic-time algorithm, the reconstruction problem can be reduced to the problem of constructing a maximum weighted matching in a complete graph consisting of the extremities of all edges in one of the initial structures. In the work presented in [
37] (Section 3), we considered the reconstruction problem on a tree with two non-leaf nodes, where the root is adjacent to arbitrarily many leaves, while a non-root non-leaf node is adjacent to exactly two leaves. There, we have described an algorithm which, for arbitrary
SCJ operation costs, solves the reconstruction problem in time of the order of
n2l2log(
nl), where
n is the number of names in the structures assigned to the leaves and
l is the number of leaves.
In this paper, we have described novel algorithms for solving the mathematical reconstruction problem for structures on the given network (
Section 2 and
Section 3), on a general tree (
Section 4) and on a specific-type tree (
Section 5). All algorithms are accompanied with proofs of their accuracy or evaluations of the maximum possible error. This publication continues the series of the mathematical papers [
21,
28,
37].
3. Algorithm for Reconstruction of Structures along a Network
3.1. Problem Setting
The concept of a structure and numerous related problems have a long history; see one of the first publications in particular [
17], and surveys [
1,
15]. Recall that a
structure [
37] is a directed graph consisting of paths and cycles, each edge of which is labeled with a name, e.g., a natural number. Here we consider the case where names are not repeated inside the structure (this is called “no paralogs”); we refer to the set of these names as the
content of the structure.
Over any structure, we consider the SCJ operations: joining extremities of edges, cutting them, inserting an isolated edge and deleting it. We confine ourselves to these operations here, although other important operations can also be considered, such as sesquialteral and double inter-merging s.
Let each leaf of the given network be uniquely assigned a loopless structure, and let positive rational numbers c1, c2, c3, and c4 be the costs of SCJ operations in the order listed above. The SCJ distance from structure a to structure b is the number of pairs of edge extremities joined in b, but not in a multiplied by c1; plus the number of pairs of extremities joined in a but not in b multiplied by c2; plus the number of edges absent in a but not in b multiplied by c3; plus the number of edges absent in b but not in a multiplied by c4. In other words, the SCJ distance is the minimum sum of costs of SCJ operations in a sequence that transforms a into b. An arrangement, R, is an unambiguous assignment of a structure to each non-leaf node of a given network, provided that the structures are already defined at the leaves.
In the reconstruction problem, a network is provided, and a structure is uniquely assigned to each of its leaves; this is required to find an arrangement R of structures over non-leaf nodes of the network at which the sum of distances between structures at the extremities of any edge is minimal. This sum is called the cost c(R) of the arrangement R. An arrangement with the minimum cost is said to be minimal.
Similar to the work presented in [
28] (Section 7), we reduce this problem to the case of equal content of all structures,
assuming that there are no loops at the leaves. To this end, loops are added at the leaves instead of edges that are absent there, i.e., if edge
z is absent in leaf
v that is present in some other leaf, then the (isolated) loop
z is added to
v. Thus, loops play the role of missing edges; and thereafter, structures with equal content are assigned to the leaves of the network. It is required to find an arrangement with the same content over the whole network, which has the minimum sum of
SCJ distances between structures along all edges of the network. Thus,
only SCJ distances and
SCJ arrangements with the same content over the whole network are considered hereafter.
An extremity of an edge has the name of this edge assigned with index 1 (if the extremity is the beginning of the edge) or with index 2 (if the extremity is the end of the edge). Denote by M a complete loopless graph consisting of the names of all extremities. Often M is viewed simply as a set, and its edges as a potential store of edges for future matchings in M; in other words, a potential edge p is any pair of extremities in M. Any structure is identified with a matching in M. A join is a pair of extremities merged in a given structure; a join has a name, a pair of names of the extremities being merged. A join in a structure corresponds to an edge in a matching, and vice versa. An arrangement of structures along a network corresponds to an arrangement of matchings along the same network; at the same time, matchings at the network nodes are defined on one and the same fixed set M. To select a matching (and, consequently, a structure) at a node, we use specially chosen weights of potential edges in M. The distance between matchings at the extremities of a network edge is taken to be the distance between the corresponding structures; then the cost of an arrangement of structures along the network is equal to the cost of the arrangement of the corresponding matchings along this network.
SCJ operations are carried over to subgraphs in M with the same cost. In M, these operations are: adding an edge between extremities not of types k1 and k2, where k is the name of the edge in the structure (join); deleting an edge between extremities not of types k1 and k2 (cut); deleting a loop between extremities of types k1 and k2 (such deleting is also called a cut); adding a loop between extremities of types k1 and k2 (such adding is also called a join). Thus, two kinds of joins with costs c1 and c4, and two kinds of cuts with costs c2 and c3, are performed over a graph G ⊆ M. An arbitrary graph in M will be called a pseudo-structure. The result of these operations applied even to a matching G ⊆ M is a pseudo-structure, but not necessarily a matching. If we pass from a pseudo-structure to the joins of initial edge extremities, we obtain a generalized notion of a “structure,” whence the term “pseudo-structure” has originated in our context.
A cyclic variant of the reconstruction problem is that all initial structures at the leaves consist of cycles, and all structures in any arrangement are also cycles. Such a cyclic structure corresponds to a complete matching on M, i.e., a matching that involves all the nodes in M.
3.2. Description of the Algorithms
Simple algorithm. Let us provide a network and, in its leaves, structures (matchings) with equal content. Then, for each potential edge
p ∈
M (i.e., a pair of extremities in the set
M) in every
leaf v of this network there is a binary
p-label (
p-attribute), which indicates whether the extremities composing
p in the matching of this
v are joined (1) or cut (0). In other words, label 1 indicates the presence of an edge
p in the
v-matching, and 0 indicates its absence in the
v-matching. We perform reconstruction along the network independently for each
p-attribute using the algorithm from
Section 2. A pair of extremities that are not joined in any leaf is reconstructed trivially. A potential edge
p ∈
M is said to be
nontrivial if the extremities that constitute
p are joined in at least one leaf of the initial data, including joins by a loop.
Then, in each node v of the network there is defined a set of all p-reconstructions, which will define a pseudo-structure G consisting of edges p ∈ M with p-label 1 in v. In other words, in each node v of the network, the p-labels at the leaves (for all p in the aggregate) uniquely define their own pseudo-structure Gv; thus, they define an arrangement G of pseudo-structures along the network. In the case of p-label 1, we will write p ∈ Gv, and in the case of p-label 0 we will write p ∉ Gv.
By successively examining extremities x ∈ M in all nodes v of the network in an arbitrary order, we remove all x-edges from the v-pseudo-structures composing G that are incidental to more than one other x-edge. Thus, we obtain a final arrangement R of structures. We call the described algorithm simple. Despite its simplicity, it has a surprising property formulated in Theorem 2.
Modified algorithm. Often one searches for an arrangement of matchings whose total number of edges (over all its matchings) is large. For this purpose, below we propose an algorithm of reasonable complexity for real computations, which we will call a modified algorithm. We cannot prove that it maximizes the specified number of edges while preserving the multiplicative exactness estimate given in Theorem 2, although our practical use of this algorithm indicates that these properties are present. The modified algorithm consists of the simple algorithm as its first stage, and two subsequent stages. We denote by R the result of the first stage, an arrangement along a given network of matchings in M. Let us describe the second and third stages of the modified algorithm.
Second stage. We successively examine all nontrivial potential edges p ∈ M. An arrangement R1 of matchings along a given network is obtained from an arrangement R of matchings in the following way. In each non-leaf node v, we add the edge p to R(v) if p ∈ Gv, or remove it if p ∉ Gv. In the resulting arrangement of pseudo-structures, we remove the original x-edge (not p itself) if at some extremity x ∈ M the incidence is violated; thus we obtain R1. For the current p, we compute the first score c(R) − c(R1), where c(·) is the cost of the arrangement.
When computing the second score, another arrangement—R1—is obtained from R as follows. We add or remove an edge p ∈ M in matchings from R according to the conditionally minimal p-attribute, which in each node v is preliminarily labeled by 0 if there is an edge in R(v) that is incidental to the extremity of the current potential edge p. Compute the second score c(R) − c(R1).
Both these arrangements—R1(p), p ∈ M—are used to compute the maxima over p of the first and second scores. Let the first of them be attained at an edge p′ with value a, and the maximum of the second score be attained at an edge p″ with value b. If a ≥ 0 and a > b, we replace R(p′) with R1(p′), and on the other edges R remains unchanged. If a = 0, the replacement is made if it strictly reduces the total number X(R) of connected components (we say, components) in all structures of the arrangement R relative to R1. If b ≥ 0 and a ≤ b, then we replace R(p″) with R1(p″) with the same stipulation if b = 0; on the other edges, R is unchanged. Otherwise, R1 = R. Obviously, the cost c(·) does not increase when passing to R1.
We repeat this iteration while one of the two things is realized: the cost c(R) or the total number of components X(R) strictly decreases when passing to the arrangement R1.
Third stage of the modified algorithm is descent to a local minimum, as described in the work presented in [
28] (Section 7). Let us recall how this descent is performed. For each node
v of a given network and for each nontrivial potential edge
p ∈
M,
denote by pno the cost
c2 or
c3 (depending on whether
p is a loop) of removing
p multiplied by the number
A of network edges entering
v at the beginnings of which
p is present, summed with the cost
c1 or
c4 (with the same dependence) of adding
p multiplied by the number
B of network edges leaving
v at the ends of which
p is present. Thus,
pno = (
c2 or
c3)·
A + (
c1 or
c4)·
B. Similarly, let
pyes denote the cost
c1 or
c4 of adding
p multiplied by the number of network edges entering
v at whose beginnings
p is absent, summed with the cost
c2 or
c3 of removing
p multiplied by the number of network edges leaving
v at whose ends
p is absent. For each node
v of the network, to each nontrivial potential edge
p ∈
M we assign the weight
pyes −
pno, and for
v we construct a minimum weighted matching in
M with these weights, which we assign to
v. In the other nodes of the network, we keep the same matchings, thus, obtaining an arrangement
R1(
v). Let the difference,
c(
R) −
c(
R1(
v)), which is maximal over all nodes
v be attained at a node
v′. If this difference is strictly positive, we replace the arrangement
R with
R1.
We repeat this iteration while c(R) − c(R1(v)) is strictly positive.
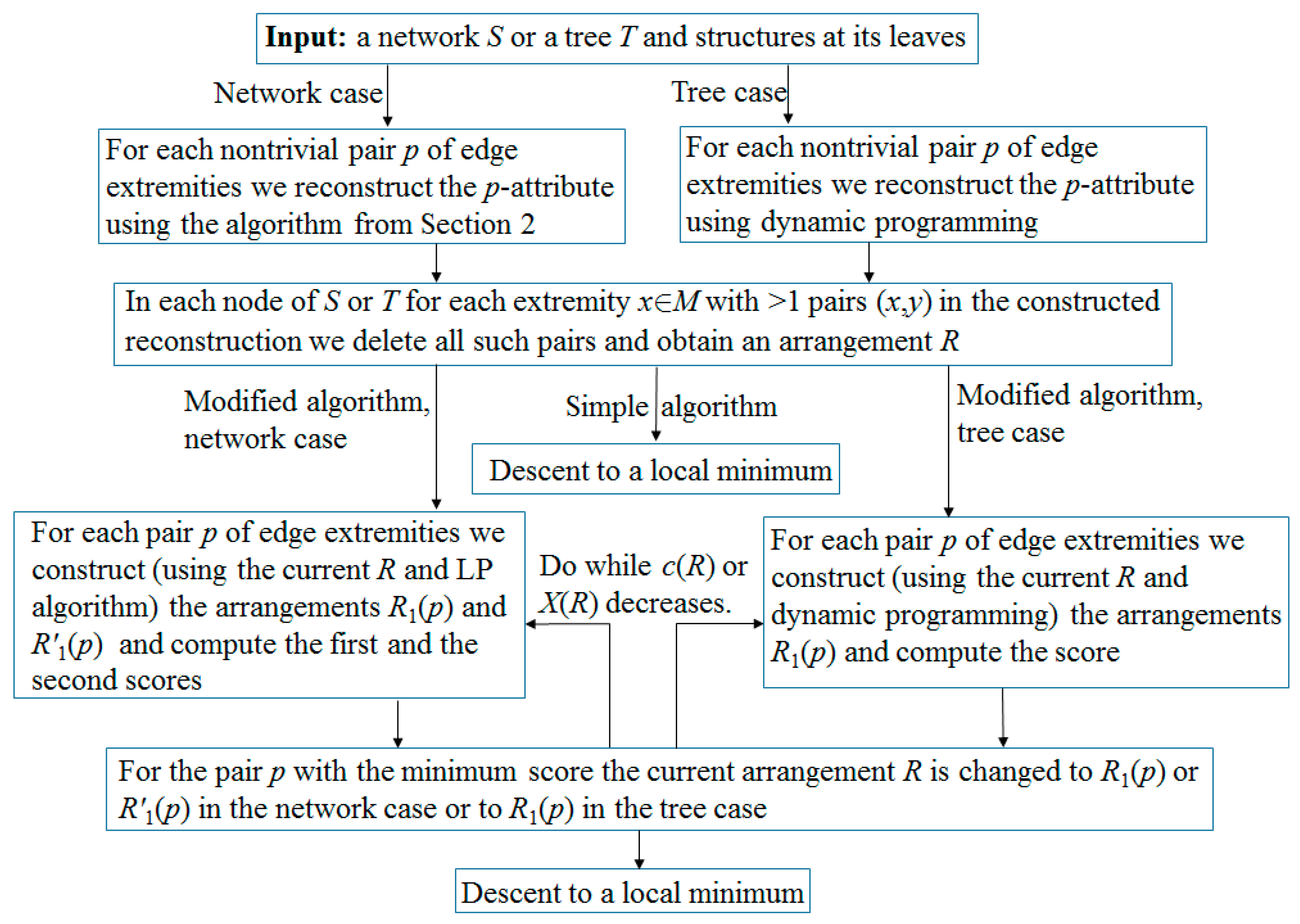
Remark 3. According to our computational experience, if the initial network is a tree, then it is more efficient to use dynamic programming instead of the above LP algorithm. Namely, the reconstruction of each p-attribute is performed by induction from leaves to root, resulting in pseudo-structures at all nodes. By removing unnecessary edges in them, we complete the simple algorithm. In the second step of the modified algorithm, instead of two p-scores, we by induction compute one score c(R) − c(R1). The arrangement R1 has the minimum cost among all arrangements that are obtained from R in the following way. In each node of the tree, one of the three rearrangements of matchings is independently performed: change nothing (empty rearrangement), remove the edge p (if it existed); or add it (if it did not exist), in which case we remove other edges incident to p. This is done by leaf-to-root induction: in every next node v, for each of its above rearrangements, in each of its child nodes v1 we choose the one of the above rearrangements in which c(v,v1) + c(v1) is minimal. Here, the first term is the distance between matchings at v and v1, and the second is the minimum cost of rearrangements along the subtree with root v1, which is known by the induction assumption. Having reached the root r of the tree, we choose a rearrangement in it with the smallest value of , where the vi are all child nodes of r. Then, by backward pass, we construct the arrangement R1 itself. The flow chart of the algorithm is shown in Figure 2. 3.3. Exactness of the Algorithm
For an arrangement of pseudo-structures along a given network, an event on an edge e of the network is that some potential edge p ∈ M enters the pseudo-structure assigned to one endpoint of e and does not enter the pseudo-structure assigned to the other endpoint of e. Recall that an edge p corresponds to a join of its extremities in the corresponding structure. An event occurs as a result of one of the four operations on the pseudo-structure, and the cost of the corresponding operation is considered to be the cost of the event. These operations are a join of extremities of pseudo-structures (with cost c4 if they are extremities of the same edge, and c1 otherwise) and a cut of extremities of pseudo-structures (with cost c3 if they are extremities of the same edge, and c2 otherwise).
The
cost of an arrangement of pseudo-structures (in particular,
structures) is the sum of costs of all events on all edges of the network. On
any edge e of the network, this sum is defined in two more equivalent ways; below, we use either of them without specifying. First, as the sum of costs
c1,
c2,
c3, and
c4 of
SCJ operations that transform a pseudo-structure at the beginning of
e into a pseudo-structure at the end of
e (see
Section 3.1); the definition for structures is just the same. Second, as the sum over all edges
p ∈
M of the costs of transitions along an edge
e of the network between
p-labels on its extremities, which (in our case) result from the reconstruction of
p-labels at the leaves.
Denote by cmin the minimum of costs of the operations and by cmax the maximum of these costs. Costs of all operations can be considered as integers, which would not lessen the generality of our presentation.
Theorem 2. (a) The simple and modified algorithms admit a multiplicative error of at most cmax/cmin. In particular, both algorithms are exact under equal costs of operations. The runtime of the simple algorithm is of the order of n·s·L, where n is the cardinality of the set M, s is the size of the given network, and L is the time required to solve the above-mentioned linear programming problem, whose size depends linearly on the size of the network. The runtime of the second stage of the modified algorithm is of the order Cns(L + s) + (ns)2·L + ns3, where C is the cost of the minimum arrangement of structures along the network. The runtime of the third stage of the modified algorithm is of the order n2·(s + log n)·sC.
(b) If a given network is a tree, then both algorithms are also exact in the case c1 = c4, c2 = c3, c2 ≥ c1.
Proof. (a) In the simple algorithm, the number of events in its final arrangement R of structures is not greater than the number of events in the arrangement G of pseudo-structures, since at each iteration the number k of events on an arbitrary network edge e with some extremity x ∈ M does not increase. Indeed, if at both ends of e in their pseudo-structures the extremity x is incident to more than one other extremity, then after an iteration all these edges are missing. If at both ends of e the extremity x is incidental to no more than one extremity, then the same edges remain at the ends. If there is, at most, one x-edge at one end of e and strictly more than one at the other end, then all x-edges at the second end are removed, and k does not increase. Let k and k′ be the numbers of events in the arrangements G and R, respectively; then k ≥ k′.
The total cost of events in G is c(G) ≥ cmin·k, and for R it is c(R) ≤ cmax·k′. The cost c(G) is minimal among the costs of all arrangements of pseudo-structures. Therefore, for the cost C of the minimal arrangement of structures, we have c(G) ≤ C. Therefore, c(R)/C ≤ cmax/cmin. At the second and third stages of the modified algorithm, the arrangement cost does not increase. Hence we obtain the first claim of the theorem. The estimate for the runtime of the simple algorithm follows from its description.
At each iteration in the second and third stages of the modified algorithm, we have
c(
R1) <
c(
R)
or X(
R1) <
X(
R). In any structure, the number of components takes a value from 1 to the integer
n/2. Hence, for the order of the runtime of the second stage, we obtain an upper estimate (
C +
ns)
·ns·(
L +
s), since this estimate is
c(
R) +
ns/2 =
C +
ns/2 multiplied by the estimate for the runtime of one iteration, which for each nontrivial pair
p (their number being of the order
ns) includes specifying the data of the LP problem, solving the problem, and processing the result. The estimate for the runtime of the third stage follows from the estimate for the time to construct a minimal matching in any graph with
n′ vertices and
m edges, which is of the order of
n′·
m + (
n′)
2·log(
n′), [
42] (Chapter 11).
(b) In the simple algorithm, first an arrangement G of pseudo-structures is formed as the union of minimal arrangements of p-labels over all p ∈ M and all nodes v of the network. Then, as a result of cuts and joins, an arrangement R of structures is output. In the simple algorithm executed on a tree, after every next arrangement, the number of cut events (i.e., those with costs c2 or c3) does not increase. Indeed, this number can only increase (and only from 0 to 1) on an edge e of the tree at the beginning of which some extremity x ∈ M is incident to exactly one extremity y and at the end x is incident to more than one extremity, possibly including y. Consider the edge e and the component K in the set consisting of the node v of the original tree in the matchings of which the extremity x is incident to more than one extremity y; the edge e enters K (from the root side). Only e enters K; leaves of the tree do not belong to K. Iteration on edges that connect nodes from K will not increase the number of events. On an edge leaving K (there are at least 2 such edges), the number of x-cut events strictly decreases. Thus, the simple algorithm does not increase the number of cuts with cost c2 = c3; assume that the algorithm reduces this number by k2 ≥ 0.
By part (a) of Theorem 2, the simple algorithm does not increase the number of all events. Therefore, for the number k1 by which the number of joins with cost c1 = c4 is increased, we have k1 ≤ k2. The condition c2 ≥ c1 implies that the simple algorithm changes c(G) to −c2·k2 + c1·k1 ≤ 0; i.e., c(G) ≥ c(R) and c(G) = c(R), since c(G) is the minimum cost. Hence, the algorithm outputs an arrangement R of structures whose cost c(R) is minimal even among all arrangements of pseudo-structures. Thus, the simple algorithm is exact, and then the modified algorithm is exact too. □
Remark 4. For the cyclic reconstruction problem we apply the following algorithm, which we will call cyclic. It differs from the modified algorithm only in the third stage, which we will now call the cyclic descent. Namely, instead of a minimum matching, the algorithm constructs a minimum complete matching and hence the structure at node v’ is always replaced by a cyclic structure. The difference c(R) − c(R1) can be negative if the structure in v’ was not cyclic before the replacement. The cyclic algorithm is heuristic; for it, the authors did not succeed in proving the error estimate formulated in Theorem 2.
Remark 5. There is another possible approach in the problem of structure reconstruction. To each nontrivial potential edge p∈ M we associate a set of the same variables at the network nodes and edges as in the LP-problem of Section 2.2, i.e., xp at the network nodes and zp at the edges. In each leaf, xp = 1 if the edge p belongs to the matching, and xp = 0 otherwise. For each p, we impose the corresponding constraints: all variables are in [0, 1], xp + yp + zp ≤ 2, xp + zp ≥ yp, yp + zp ≥ xp, and xp + yp ≥ zp. We impose new constraints: for each non-leaf node of the network and for each extremity x∈ M we have . The objective function is the sum of the objective functions of the LP problem mentioned above: If the solution of this LP problem is Boolean, then at each node of the network it defines a matching in M. However, the solution of this LP problem need not necessarily be Boolean, and then the coordinates of the solution can be viewed as probabilities for the edge p to enter the desired matching.
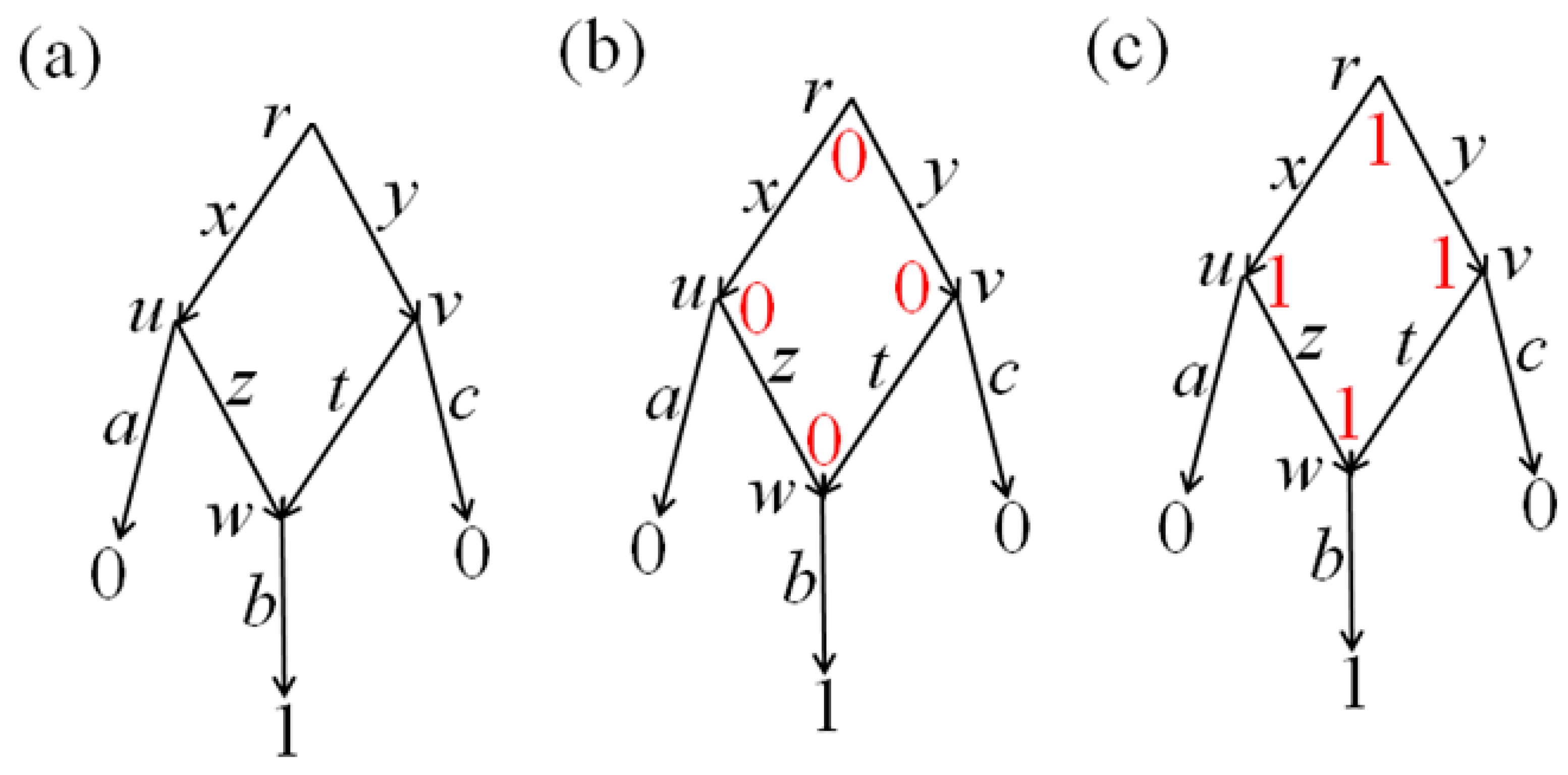
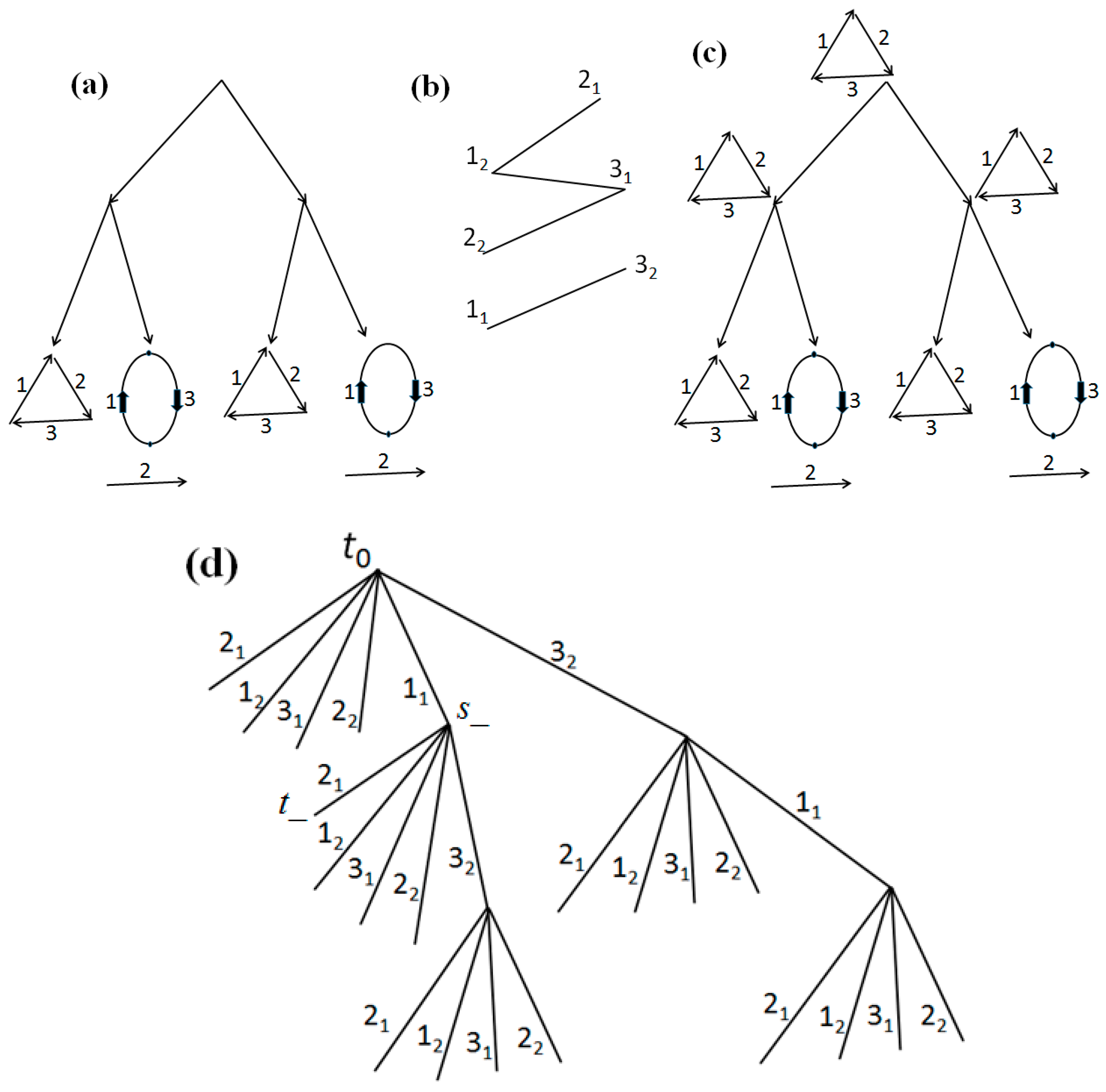
3.4. Example of the Algorithm Operation
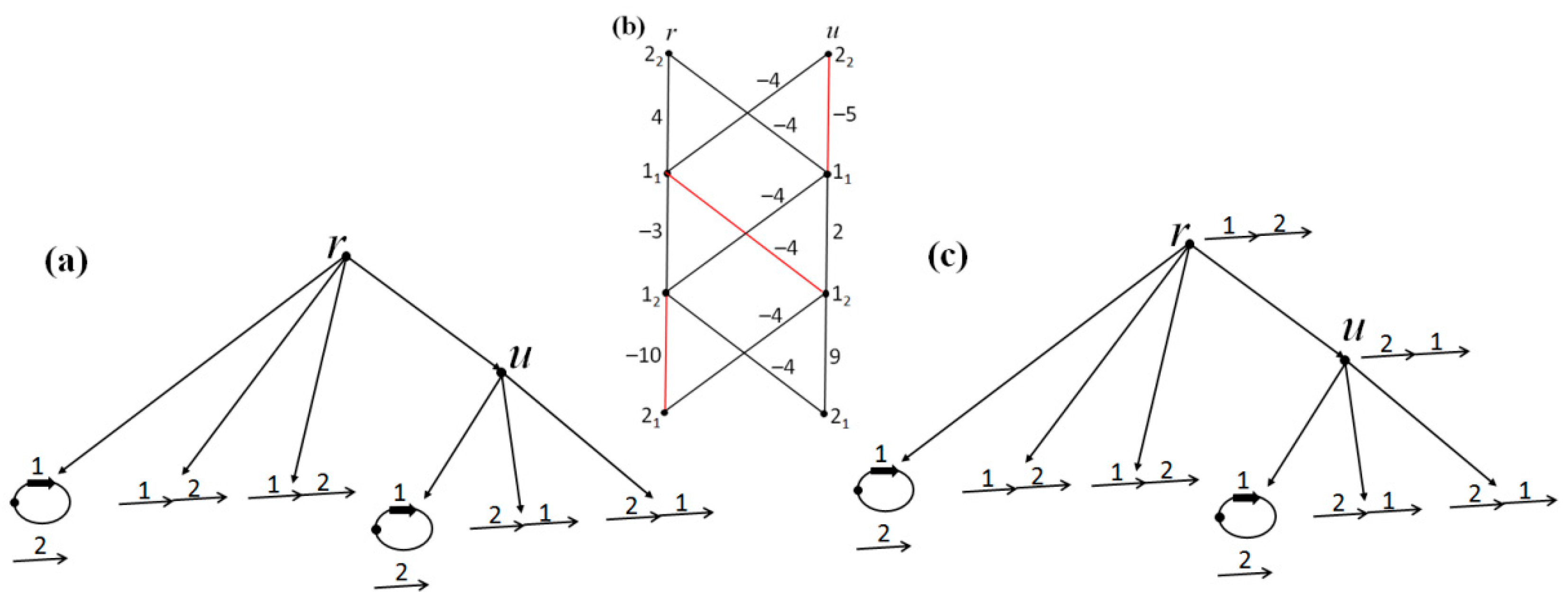
Consider the network with the initial data shown in its leaves in
Figure 3a. Let
c1 =
c2 =
c3 =
c4 = 1. Applying the algorithm of
Section 2 to the eight joins present in the leaves, we obtain that only the edge (1
2,2
1) is present in the arrangement
G of pseudo-structures shown at the non-leaf nodes of the network. Thus,
G is already an arrangement of structures, which is shown in
Figure 3b and is minimal. Its cost is 8.
Now let
c1 =
c4 = 6 and
c2 =
c3 = 1 (i.e., let a join of extremities have cost 6 and a cut of extremities have cost 1). The arrangement
G of pseudo-structures is the same at all nodes and is shown in
Figure 4a. Let us apply the modified algorithm. Let the order of examining the extremities
x at the first stage be 2
1, 3
1, 3
2 (here the final result does not depend on the choice of the order). After the first step, only the edge (1
1,2
2) remains in
G. At the second stage and at the first iteration, the maximum of the two scores (the first one) attains the maximum of eleven on the edge (1
2,2
1), at the second iteration it attains the maximum of 0 on the edge (1
1,3
2), and at the third iteration it attains the maximum of four on the edge (2
2,3
1). After adding these edges and removing the edge (1
1,2
2), we obtain the resulting arrangement,
Figure 4b. This arrangement is minimal with cost 35.
4. Reconstruction of Structures along a Tree
4.1. Problem Setting
We again consider the same problem of reconstructing structures, now not along a network, but along a given tree
T, which in the general case is nonbinary (sometimes referred to as polytomous). As in
Section 3, we first ensure equal content of structures defined at leaves, and therefore, equal content of all desired structures at non-leaf nodes of the tree. As above, the reconstruction problem reduces to constructing in each non-leaf node of the tree a matching on the same set
M of names of extremities of all edges at the leaves. In contrast to Theorem 2, we describe here another algorithm for solving this problem based on dynamic programming, which turns out to be exact if a natural condition formulated below is satisfied. It is important that the runtime of the algorithm is estimated by a third-order quantity with respect to the size of the original problem multiplied by the size of the auxiliary tree, which usually takes small values.
For an extremity x ∈ M, by an x-arrangement we call an unambiguous assignment of at most one edge p ∈ M incident to x (in the cyclic variant, exactly one edge) to each node of a given tree T. An x-arrangement is a particular case of an arrangement of matchings. The cost of an x-arrangement is defined to be its cost as of an arrangement of matchings; in this case, only edges incident to x are left in the leaves of T.
An x-arrangement with a minimum cost is constructed by induction. At leaves, an x-arrangement is given by an edge with which the extremity x is joined, or by an empty set. An x-edge is defined to be either an edge in M with extremity x or the symbol Ø of the empty set. For a fixed extremity x ∈ M, we examine the nodes of T from leaves to the root; at each node v and for each potential x-edge p in v, we compute the function c(v,p) = åv′ c(v′). Here, c(v,v′,q) is the number of events on the edge (v,v′) if q is assigned to a child node v′ of the tree, c(v′,q) is the minimum cost of an x-arrangement over a subtree with root v′ in which the x-arrangement is q, and c(v′) = minq c(v,v′,q) + c(v′,q). Let the minimum of c(v′) be attained at q(p); c(v,p) and q(p) are stored in v. At the root r ∈ T, we choose the x-edge with the minimum value of c(r,p) over p, and then perform the backward pass of dynamic programming. The resulting x-arrangement has the minimum cost among all x-arrangements and is said to be minimal.
Let an arrangement R of matchings along T be given. Then, a conditionally minimal x-arrangement R′ along T extending R is defined as the minimum-cost x-arrangement among all x-arrangements containing all x-edges from R and not containing x-edges whose other extremity is incident to an edge from R. It is constructed by dynamic programming similarly to the preceding algorithm. Note that all x-edges from R are included in R′; in this sense, R′ is an extension of R.
On the other hand, let us be given an arrangement
R of matchings along
T and a set
X ⊆
M. By a
conditionally minimal extension G of the arrangement
R with respect to
X, we call an extension of
R by edges of conditionally minimal arrangements of
p-labels along nontrivial potential edges
p ∈
M one or both extremities of which are not contained in
X. The condition is that a
p-label at node
v ∈
T is 1 if
p ∈
R(
v), and is 0 if
p is incident to an edge
q ∈
R(
v),
q ≠ p. Unlike
R,
G is an arrangement of pseudo-structures along
T, and to construct it, we use the algorithm provided in
Section 2.2; see also Remark 1 in
Section 2.3. Obviously,
R ⊆
G.
We will need a separate auxiliary tree L with root t0, which represents variants of possible extensions of the empty arrangement along the tree T. A path in L from the root to a leaf shows one variant of such a consecutive extension of the empty arrangement. Throughout what follows, by a path in L we mean a path from t0, which is not specified explicitly. For each edge l ∈ L, we denote its beginning by l+ and its end by l−. Each node l– will be assigned with an extremity x ∈ M, which we denote by x(l). Moreover, in any path to a node l– ∈ L, the extremity x(l) occurs only once, at the very end. From any non-leaf node in L there emerges edges uniquely labeled by x(l), for every x ∈ M that does not occur in a path to l+, see Figure 6d.
4.2. Description of the Algorithm
Recall that the cost of an arrangement R of structures or an arrangement G of pseudo-structures along a tree T is denoted by c(R) and c(G).
Before each iteration, the algorithm has a current tree
L with some leaves marked as
dead-end leaves. Moreover, each node
l− ∈
L is unambiguously assigned with an arrangement
R of matchings and an arrangement
G of pseudo-structures, both along the initial tree
T (in the leaves of which, initial structures are still fixed; they are also pseudo-structures). Even before each iteration, the algorithm has an
arrangement U of matchings at non-leaf nodes in
T (at the leaves it coincides with the original structures) and a cost
u of this arrangement. At each iteration except the last one, the tree
L is continued “downwards”, see the first example in
Section 4.4.
Before the first iteration, L consists of a single non-dead-end root t0 ∈ L, the arrangement R(t0) along T consists of empty matchings (at leaves there are original matchings), G(t0) is the conditionally minimal extension of the arrangement R(t0) with respect to the empty set, i.e., the union of the minimal arrangements of all p-attributes. Finally, U is the empty arrangement (except for the leaves, which contain the original matchings), and u = c(U).
Let us describe the iteration itself. Find a non-dead-end leaf
s− ∈
L (at the first iteration,
s− =
t0) with the minimum (over all non-dead-end leaves) value of the cost
c(
G(
s−)). Extend
L with all edges
l outgoing from
s− with all labels
x(
l) that do not occur on the path to
s–. For each added edge
l ∈
L, construct an arrangement
R(
l−) of matchings and an arrangement
G(
l−) of pseudo-structures, both along
T, see the first example in
Section 4.4:
- (1)
R(l−) is the union of the arrangement R(s−) and the conditionally minimal x(l)-arrangement that extends R(s−), where l+ = s−;
- (2)
G(l−) is the conditionally minimal extension of the already obtained arrangement R(l−) with respect to X(l) ⊆ M. Here, X(l) is the set of extremities that occur on the path to l−.
If c(G(l−)) ≥ u or G(l−) is already an arrangement of matchings, then l− is marked as dead-end. Over all added nodes l− (outgoing from s−), we find the minimum c′ of costs c(R(l−)), and if G(l–) consists of structures, then including also the costs c(G(l−)). Let c− be attained at t–. If c′ < u, then the former price u is changed to c’ and the former arrangement U is changed to either R(t−) or G(t−) (according to which of them the minimum is attained). The ambiguity that may arise in this case is eliminated by using a random choice, which is the end of the iteration.
The iterations are performed until all the leaves in the next tree L are marked as dead-end. Then, as the final arrangement along T, the algorithm outputs the current arrangement U and its cost u.
The cyclic variant of the algorithm contains an additional step executed after all the above iterations. Namely, if the final arrangement is not cyclic, then, by examining the extremities x ∈ M in an arbitrary order, we extend the current arrangement by a conditionally minimal x-arrangement with the following condition: at each node v ∈ T the matching property is not violated, and the extremity x enters the matching. This condition can always be ensured, since the current arrangement at each node covers an even number of extremities and n = |M| is an even number. Indeed, if an x-edge is missing, it can be chosen without violating the matching property.
The flow chart of the algorithm is shown in
Figure 5.
4.3. Justification of the Algorithm
The following property is often fulfilled in structure reconstruction problems arising, for example, in evolutionary genomics, cell type construction, etc.
Extremity enumeration property. There exists a bijective enumeration of all extremities r1, …, rn in M for which the arrangement Rn of matchings is minimal and for which R1 is the union of R(t0) and the unique minimal r1-arrangement, R2 is the union of R1 and the unique conditionally minimal r2-arrangement extending R1, etc. Thus, each Ri+1 is an extension of the preceding Ri.
Both the fulfillment and non-fulfillment of the enumeration property depend on a particular enumeration, as can be seen in the examples in
Section 4.4. In the first one, this property is satisfied for the enumeration of extremities 2
12
23
13
21
11
2 and is not satisfied for the enumeration 1
11
22
12
23
13
2, since in the latter case the uniqueness is violated at
R2. In the second example in
Section 4.4, any enumeration obeys the enumeration property. We emphasize that the algorithm presented below does not use any initial enumeration of extremities in
M.
Theorem 3. If the extremity enumeration property is fulfilled in the reconstruction problem, then the above algorithm produces an exact solution. Its runtime is of the order of n·s2·l, where n is the cardinality of the set M, s is the size of the tree T, and l is the size of the resulting tree L.
Proof. Let r1, …, ri, …, rn be an enumeration of all extremities satisfying the above property, and let Ri be a current arrangement of matchings. In the final tree L, consider a path with labels r1, r2, …, ri from the root to a dead-end leaf. By induction, due to the uniqueness of conditionally minimal ri-arrangements, we have R(ri) = Ri.
Let us check that c(G(ri)) ≤ c(Rn). Consider potential edges p = (x,y) ∈ M for which x,y ∈ X(ri) is satisfied; denote the set of such edges by I, and denote its complement by I’. Then, we have
and , where S′p and Sp are the arrangements of p-labels induced by G(ri) and Rn, respectively. The arrangements G(ri) and Rn are obtained from R(ri) = Ri by adding the edges p ∈ I’. Therefore, G(ri) and Rn differ at edges from I’ only; i.e., the first terms in these sums are the same. Since in G(ri) for p ∈ I’ every arrangement of p-labels is minimal, we obtain c(G(ri)) ≤ c(Rn).
Consider two cases in which ri is a dead-end leaf.
Let G(ri) be an arrangement of structures. Then, c(G(ri)) = c(Rn) = C, where C is the minimum cost of all arrangements of structures along T. For edges from ri–1 (from t0 for i = 1) we have c′ = C, and if c′ < u, then at this iteration we let u = C and U = G(ri), and at the next iterations these u and U cannot change. The algorithm outputs the final arrangement U with cost C. If C = u, then the algorithm had obtained the final arrangement U with cost C at the previous iteration.
Now, let the first case c(G(ri)) ≥ u, occur, where u is computed at the previous iteration. Then, c(G(ri)) ≥ u ≥ c(Rn) and c(G(ri)) = c(Rn) = C = u = c(U). Therefore, the algorithm does not change u and U at this and the next iterations, outputting the final arrangement U with cost C.
The estimate for the runtime of our algorithm follows from the fact that for each node in L, the auxiliary dynamic programming algorithm that constructs an arrangement of p-labels is executed of the order of ns times, and each time its runtime is of the order of s. □
4.4. Examples of the Algorithm Operation
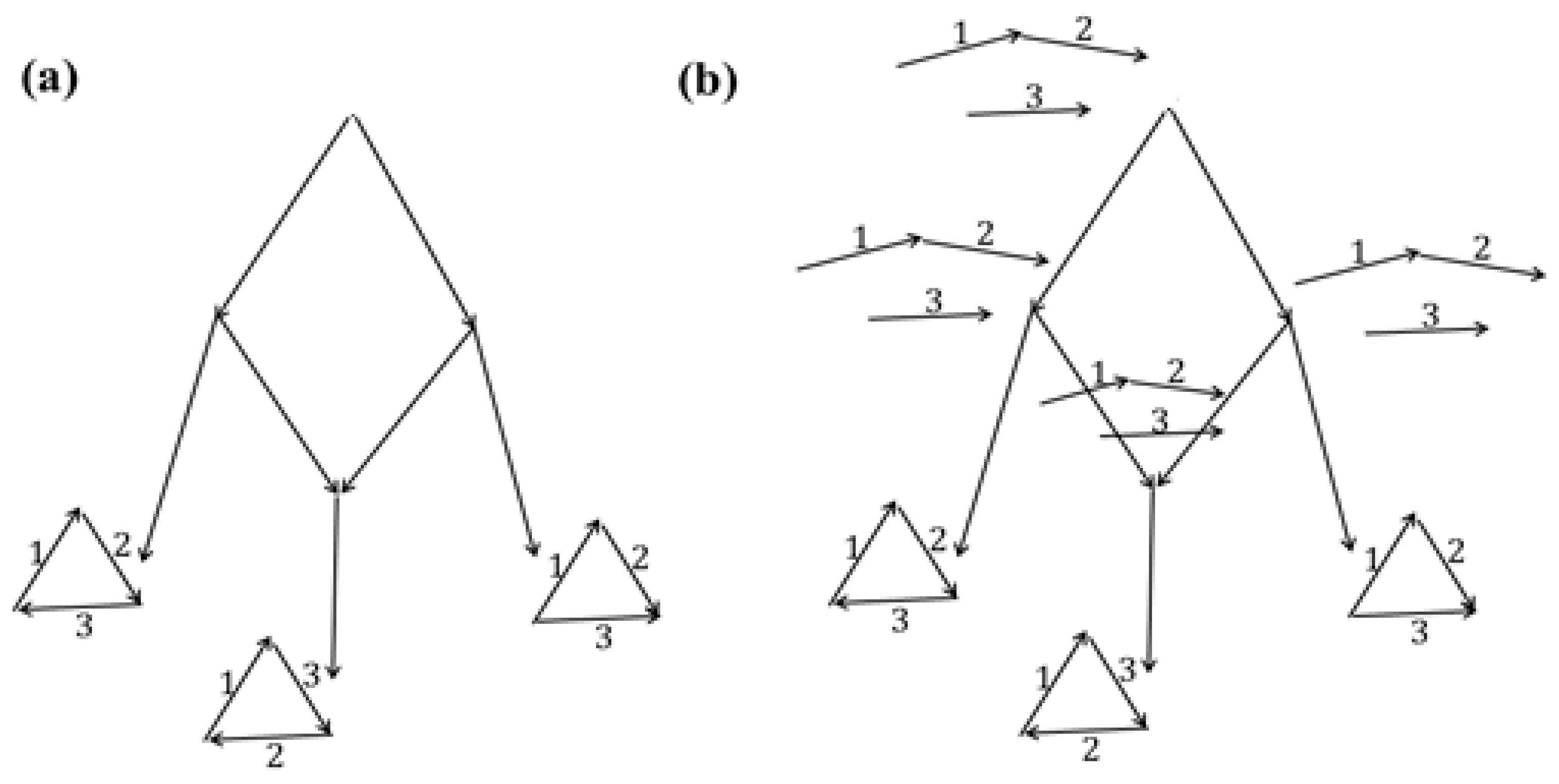
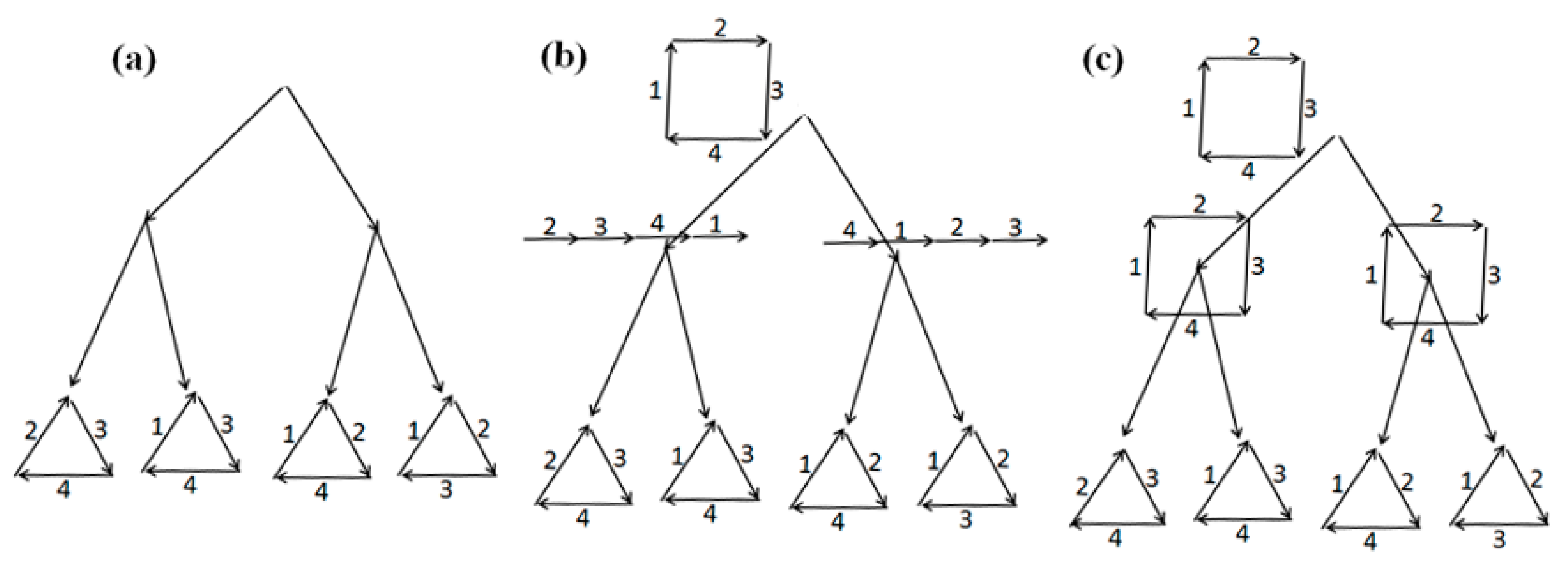
First example. Consider a tree
T with the structures at its leaves shown in
Figure 6a. Let costs of joins be
c1 =
c4 = 3 and costs of cuts be
c2 =
c3 = 2. First, the tree
L consists of the root
t0. The arrangement
R(
t0) of matchings is the same at all non-leaf nodes in
T (empty one, except for the leaves), and
u =
c(
U) = 30. And the arrangement
G(
t0) of pseudo-structures with cost 12 is the same at all non-leaf nodes in
T (the original one at the leaves) and is shown in
Figure 6b.
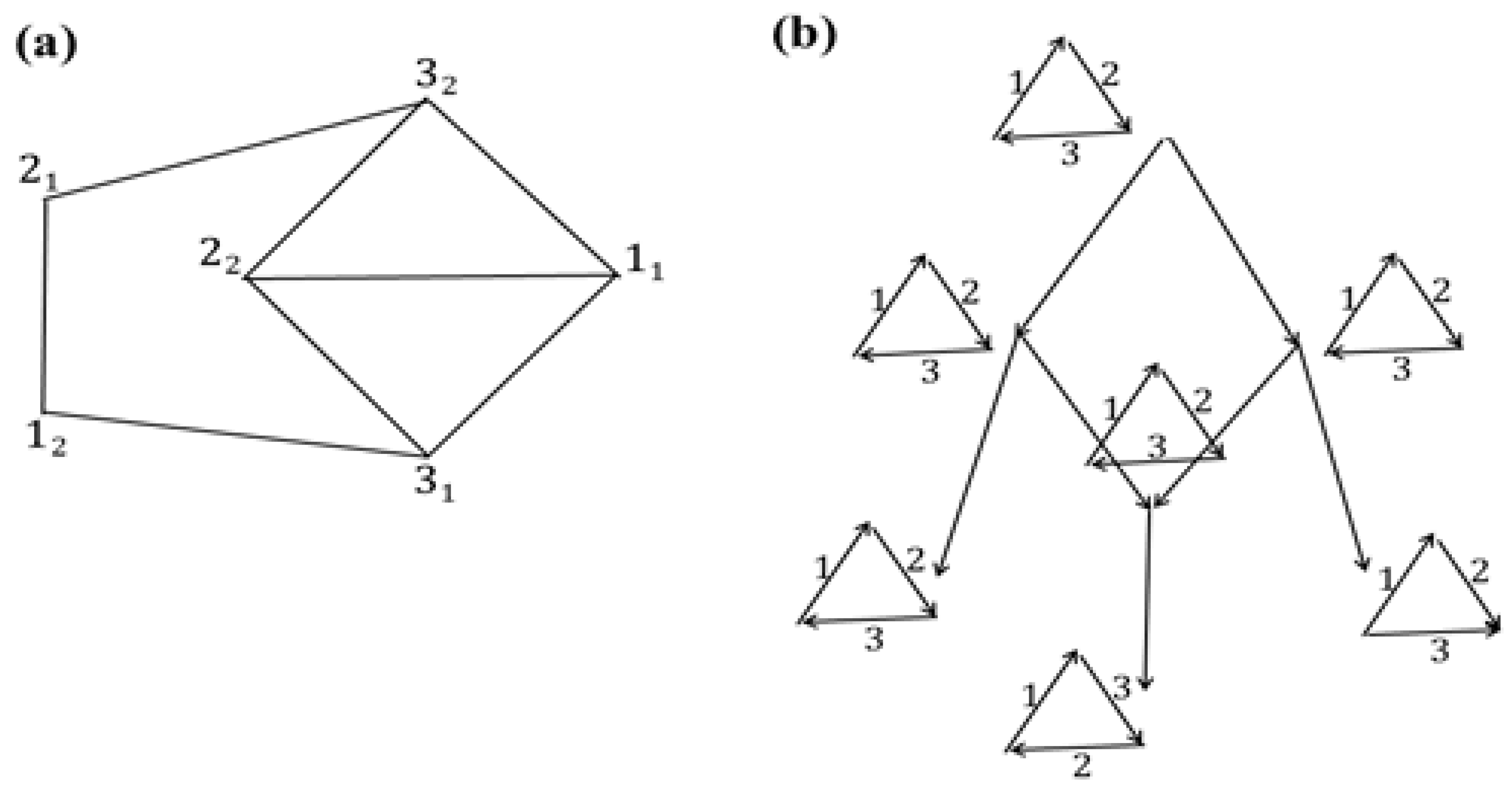
Let us extend the tree
L with edges outgoing from
t0; then we obtain that for the edge
l with end
l– = 2
1 we have the following:
R(2
1) = (1
2,2
1) is the same at all non-leaf nodes in
T,
c(
R(2
1)) = 28, and similarly the arrangement
G(2
1) is the same at all non-leaf nodes in
T and coincides at them with the matching {(2
2,3
1), (1
2,2
1), (1
1,3
2)} shown in
Figure 6c. Therefore, the node 2
1 is marked as dead-end. Here,
c(
G(2
1)) = 14. For other new edges
l ∈
L, we obtain costs of the structures
R(
l–) and
G(
l–) not less than 14:
R(1
2) = (1
2,2
1) or
R(1
2) = (1
2,3
1),
c(
R(1
2)) = 28, and
G(1
2) = {(2
2,3
1),(1
2,2
1),(1
1,3
2)} with
c(
G(1
2)) = 14 or
G(1
2) = {(1
1,3
2),(1
2,2
1)} with
c(
G(1
2)) = 16. Next,
R(3
1) = (1
2,3
1) or
R(3
1) = (2
2,3
1), in both cases
c(
R(3
1)) = 28; and
G(3
1) =
G(1
2). Then,
R(2
2) = (2
2,3
1),
c(
R(2
2)) = 28,
G(2
2) =
G(2
1); and
R(1
1) = (1
1,3
2),
c(
R(1
1)) = 18,
G(1
1) =
G(
t0); finally,
R(3
2) =
R(1
1),
G(3
2) =
G(
t0). Ends of the edges from
t0 are dead-end except for 1
1 and 3
2. Therefore,
c’ = 14 and
t– = 2
1; hence we obtain
U =
G(2
1) and
u = 14.
Further extension of
L does not change the values of
U and
u: for instance, for the end of the path
t01
11
2 (
l– = 1
2) we have
R(1
2) = {(1
1,3
2), (1
2,2
1)} at all non-leaf nodes
v ∈
T, or alternatively
R(1
2) = {(1
1,3
2), (1
2,3
1)}; both arrangements have cost 16. Next,
G(1
2) = {(2
2,3
1), (1
2,2
1), (1
1,3
2)} at non-leaf nodes
v ∈
T, or alternatively
G(1
2) = {(1
1,3
2), (1
2,3
1)}; these are arrangements with costs 14 and 16, respectively. For other edges
l ∈
L from the node 1
1 of the tree
L, we obtain that the costs of arrangements of the structures
R(
l–) and
G(
l–) are at least 14:
R(2
1) = {(1
1,3
2), (1
2,2
1)},
c(
R(2
1)) = 16,
G(2
1) = {(2
2,3
1), (1
2,2
1), (1
1,3
2)},
c(
G(2
1)) = 14;
R(3
1) = {(1
1,3
2), (1
2,3
1)} or {(1
1,3
2), (2
2,3
1)},
c(
R(3
1)) = 16,
G(3
1) =
G(1
2);
R(2
2) = {(1
1,3
2), (2
2,3
1)},
c(
R(2
2)) = 16,
G(2
2) =
G(2
1);
R(3
2) = (1
1,3
2),
c(
R(3
2)) = 18,
G(3
2) =
G(
t0). Ends of the edges from 1
1 are marked as dead-ends except for 3
2. Thus, the algorithm outputs the same
U as an arrangement and the same
u as its cost until all nodes in a current
L become dead-ends. Such a final tree is shown in
Figure 6d, in which all leaves correspond to the extremities 2
1, 1
2, 3
1, 2
2 in
M. The algorithm outputs the previously obtained
U and
u as the final ones.
It is easy to directly check that the arrangement U has the minimum cost u = 14, i.e., it is minimal.
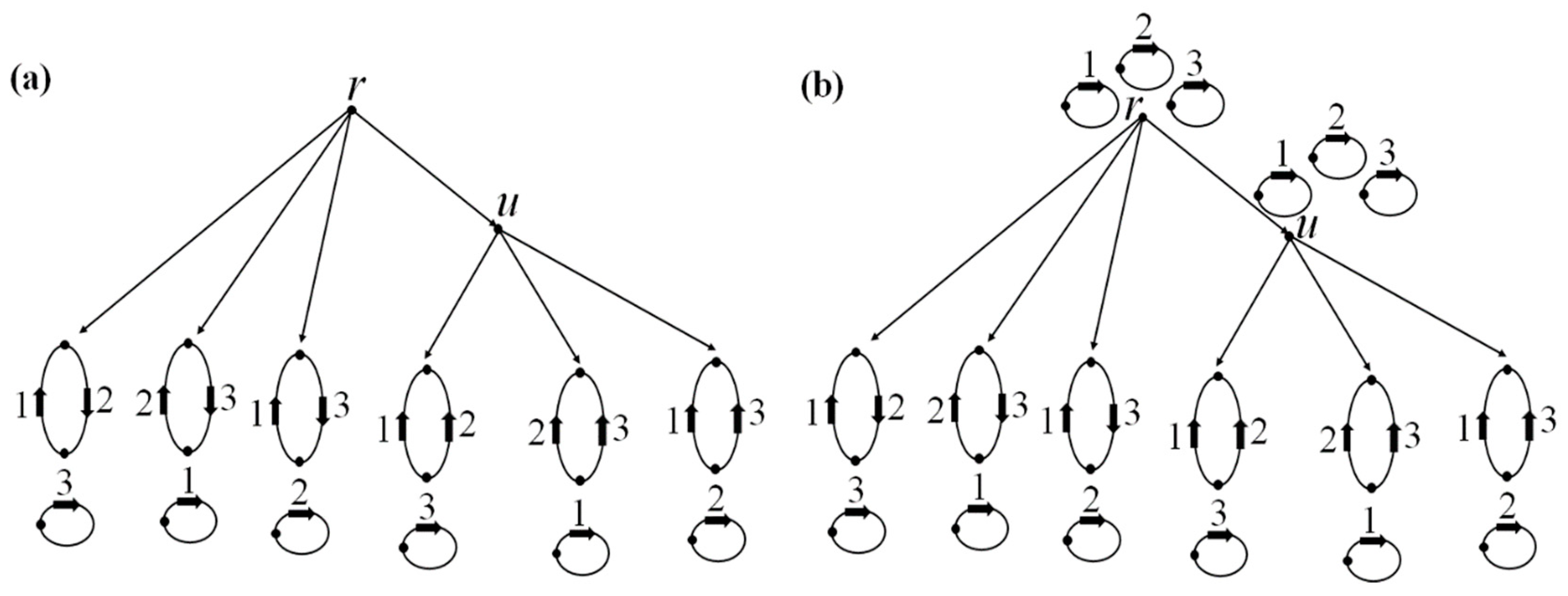
Second example. Consider a tree
T with the structures at its leaves shown in
Figure 7a; edges missing at the leaves are replaced with loops, which is not shown in this figure. Let the operation costs be the same as in the first example. If a nontrivial edge
p ∈
M is present in exactly one leaf, then
p-labels at all non-leaf nodes are 0. If
p is present in exactly two neighboring leaves, then the
p-label at two non-leaf nodes is 1 and at the third is 0. If
p is present in exactly two non-neighboring leaves, then the
p-label is 1 at all non-leaf nodes. Hence we see that the arrangement
G(
t0) of pseudo-structures consists of structures, and the algorithm outputs it as the final one,
Figure 7b. The resulting tree
L consists of a single dead-end root
t0.
In the cyclic version of the algorithm with the same costs, the additional step adds one edge at each of the two non-root nodes. The final arrangement is shown in
Figure 7c.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









