
A Novel Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes


Abstract
1. Introduction
1.1. Literature Review
1.2. Brief Introduction of a Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set
2. Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes
2.1. Problem Definitions
2.2. Similarity Definitions with Membership Degree Based on the AFS
2.3. Bi-Similarity Criterion Based on the Similarity Matrices
| Algorithm 1 (Node Deletion) |
| Input: and , the two fuzzy partition matrices of the gene expression data matrix, and , the two scale factors of the column and row for the bi-clusters to be found. |
| Output: , an (αβ)-bi-cluster that is a sub-matrix of with row set and column set with the dissimilarity scores no larger than and , respectively. |
| Initialization: and are initialized to the gene and condition sets in the gene expression data, and ; a reference gene is given by the user; the maximum acceptable dissimilarity scores of the column and row: , . |
| Iteration: |
| (1). Calculate for all , for all , and , . If and , return . |
| (2). Find column with largest
|
| Algorithm 2 (Node Addition) |
| Input: , a sub-matrix of real numbers; and signifying an (αβ)-bi-cluster. |
| Output: , , and such that and with the property that
|
| Iteration: |
| (1). Compute for all , recompute and , and add the columns if .
|
| (2). Compute for all , recompute the and , and add the rows if |
| (3). If nothing is added in the iterate, return the final and as and . |
2.4. Selection of the Reference Genes
3. Performance Indexes
3.1. Variance Index
3.2. Mean Fluctuation Degree Index
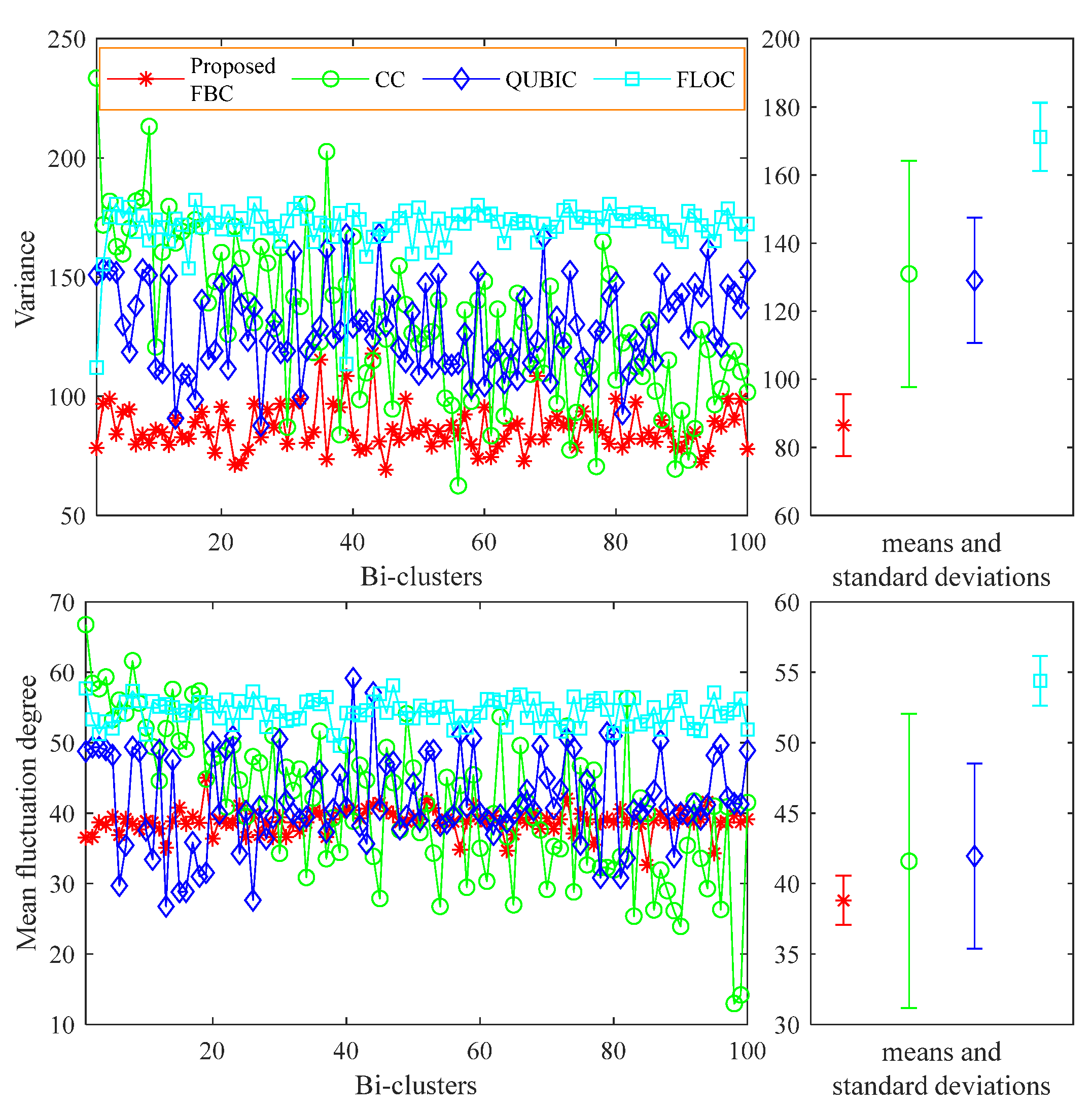
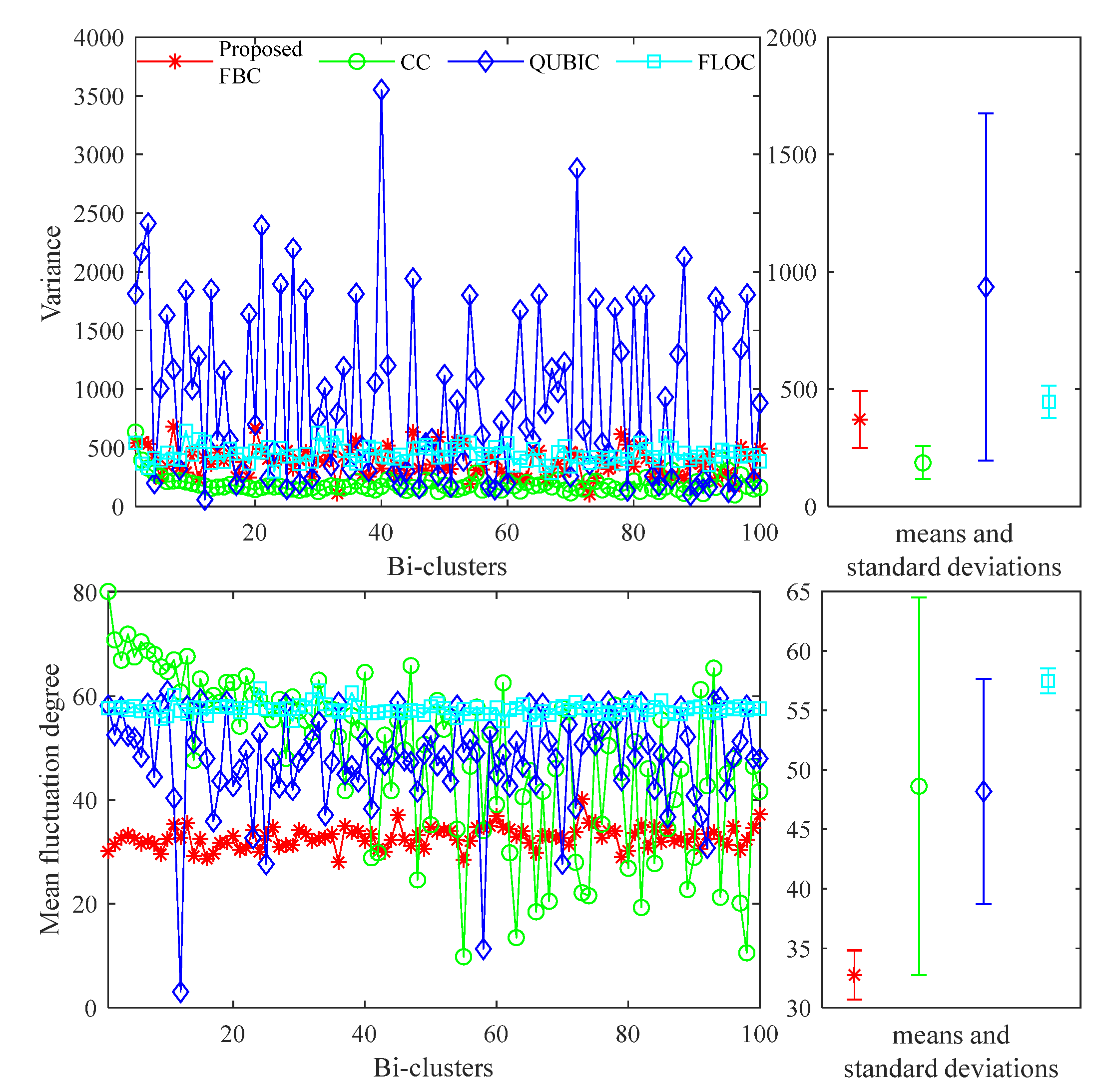
4. Experimental Studies
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dhaeseleer, P. How does gene expression clustering work? Nat. Biotechnol. 2005, 23, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Pattini, L.; Sassi, R.; Cerutti, S. Dissecting heart failure through the multiscale approach of systems medicine. IEEE Trans. Biomed. Eng. 2014, 61, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 2018, 36, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Shaw, K.; Mishra, S. Gene expression network discovery: A pattern based biclustering approach. In Proceedings of the 2011 International Conference on Communication, Computing & Security, ACM, Rourkela, Odisha, India, 12–14 February 2011; pp. 307–312. [Google Scholar]
- Yang, J.; Wang, H.; Wang, W. Enhanced biclustering on expression data. In Proceedings of the Third IEEE Symposium on Bioinformatics and Bioengineering, Bethesda, MD, USA, 10–12 March 2003; pp. 321–327. [Google Scholar]
- Cheng, Y.; Church, G.M. Biclustering of expression data. In Proceedings of the Conference on Intelligent Systems for Molecular Biology (ISM), San Diego, CA, USA, 19–23 August 2000; pp. 93–103. [Google Scholar]
- Yang, J.; Wang, H.; Wang, W.; Yu, P.S. An improved biclustering method for analyzing gene expression profiles. Int. J. Artif. Intell. Tools 2005, 14, 771–789. [Google Scholar] [CrossRef]
- Lazzeroni, L.; Owen, A. Plaid models for gene expression data. Stat. Sin. 2000, 12, 61–86. [Google Scholar]
- Ben-Dor, A.; Chor, B.; Karp, R.; Yakhini, Z. Discovering local structure in gene expression data: The order-preserving submatrix problem. J. Comput. Biol. 2003, 10, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Ihmels, J.; Barkai, N. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2003, 67, 031902. [Google Scholar] [CrossRef]
- Murali, T.M.; Kasif, S. Extracting conserved gene expression motifs from gene expression data. In Proceedings of the Pacific Symposium on Biocomputing, Lihue, HI, USA, 3–7 January 2003; pp. 77–88. [Google Scholar]
- Prelić, A.; Bleuler, S.; Zimmermann, P.; Wille, A.; Bühlmann, P.; Gruissem, W.; Hennig, L.; Thiele, L.; Zitzler, E. A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics 2006, 22, 1122–1129. [Google Scholar] [CrossRef]
- Gao, C.; McDowell, I.C.; Zhao, S. Context specific and differential gene co-expression networks via Bayesian biclustering. Comput. Biol. 2016, 12, e1004791. [Google Scholar] [CrossRef]
- Liu, X.; Wang, L. Computing the maximum similarity bi-clusters of gene expression data. Bioinformatics 2006, 23, 50–56. [Google Scholar] [CrossRef]
- Li, G.; Ma, Q.; Tang, H.; Paterson, A.H.; Xu, Y. QUBIC: A qualitative biclustering algorithm for analyses of gene expression data. Nucleic Acids Res. 2009, 37, e101. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, M.P.; Saravana, K.E. A survey on biclustering. Int. J. Innov. Res. Sci. Technol. 2016, 3, 2349–6010. [Google Scholar]
- Khalid, B.; Allab, K. Bi-clustering continuous data with self-organizing map. Neural Comput. Appl. 2013, 22, 1551–1562. [Google Scholar]
- Liu, X.; Jia, W.; Wang, Y.; Guo, H.; Ren, Y.; Li, Z. Knowledge discovery and semantic learning in the framework of axiomatic fuzzy set theory. WIREs Data Min. Knowl. Discov. 2018, 8, 1268–1292. [Google Scholar] [CrossRef]
- Lian, C.; Ruan, S.; Denoeux, T.; Li, H.; Vera, P. Spatial evidential clustering with adaptive distance metric for tumor segmentation in FDG-PET images. IEEE Trans. Biomed. Eng. 2018, 65, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Pedrycz, W.; Li, Z.; Nie, W. Constructing a virtual space for enhancing the classification performance of Fuzzy clustering. IEEE Trans. Fuzzy Syst. 2018, 27, 1779–1792. [Google Scholar] [CrossRef]
- Xu, K.J.; Pedrycz, W.; Li, Z.W.; Nie, W.K. High-accuracy signal subspace separation algorithm based on gaussian kernel. IEEE Trans. Ind. Electron. 2019, 66, 491–499. [Google Scholar] [CrossRef]
- Madeira, S.C.; Oliveira, A.L. Biclustering algorithms for biological data analysis: A survey. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2004, 1, 24–45. [Google Scholar] [CrossRef]
- Ren, Y.; Song, M.; Liu, X. New approaches to the fuzzy clustering via AFS theory. Int. J. Inf. Syst. Sci. 2007, 3, 307–325. [Google Scholar]
- Stanev, D.; Moustakas, K. Simulation of constrained musculoskeletal systems in task space. IEEE Trans. Biomed. Eng. 2017, 65, 307–318. [Google Scholar] [CrossRef]
- Li, X.; Wong, K.-C. Evolutionary multiobjective clustering and its applications to patient stratification. IEEE Trans. Cybern. 2019, 49, 1680–1693. [Google Scholar] [CrossRef] [PubMed]
- Shrimankar, D.D.; Durge, A.R.; Sawarkar, A.D. Heuristic analysis of genomic sequence processing models for high efficiency prediction: A statistical perspective. Curr. Genom. 2022, 23, 299–317. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Datasets | Yeast | Gordon-2002 | |
|---|---|---|---|
| Number of | Genes | 2884 | 1626 |
| Conditions | 17 | 181 | |
| Threshold of MSR | 300 | 3000 | |
| α | 5.0 | 5.5 | |
| β | 1.8 | 3.0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Wang, Y. A Novel Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes. Mathematics 2024, 12, 1659. https://doi.org/10.3390/math12111659
Xu K, Wang Y. A Novel Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes. Mathematics. 2024; 12(11):1659. https://doi.org/10.3390/math12111659
Chicago/Turabian StyleXu, Kaijie, and Yixi Wang. 2024. "A Novel Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes" Mathematics 12, no. 11: 1659. https://doi.org/10.3390/math12111659
APA StyleXu, K., & Wang, Y. (2024). A Novel Fuzzy Bi-Clustering Algorithm with Axiomatic Fuzzy Set for Identification of Co-Regulated Genes. Mathematics, 12(11), 1659. https://doi.org/10.3390/math12111659