
LightGBM-LncLoc: A LightGBM-Based Computational Predictor for Recognizing Long Non-Coding RNA Subcellular Localization

Abstract
1. Introduction
2. Methods
2.1. Dataset
2.2. Methodology
2.3. Feature Selection
2.3.1. PSTNPSSMC
2.3.2. RCKmer
2.4. LightGBM
2.5. Validation and Metrics
3. Results and Discussion
3.1. Feature Combination Optimization
3.2. Selection of Learning Algorithms
3.3. Parameter Optimization
3.4. Comparison with State-of-the-Art Methods
3.5. WebServer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.-X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA–disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Kitagawa, K.; Kotake, Y.; Niida, H.; Ohhata, T. Cell cycle regulation by long non-coding RNAs. Cell. Mol. Life Sci. 2013, 70, 4785–4794. [Google Scholar] [CrossRef]
- Brazão, T.F.; Johnson, J.S.; Müller, J.; Heger, A.; Ponting, C.P.; Tybulewicz, V.L. Long noncoding RNAs in B-cell development and activation. Blood J. Am. Soc. Hematol. 2016, 128, e10–e19. [Google Scholar] [CrossRef] [PubMed]
- Delas, M.J.; Sabin, L.R.; Dolzhenko, E.; Knott, S.R.; Munera Maravilla, E.; Jackson, B.T.; Wild, S.A.; Kovacevic, T.; Stork, E.M.; Zhou, M.; et al. lncRNA requirements for mouse acute myeloid leukemia and normal differentiation. eLife 2017, 6, e25607. [Google Scholar] [CrossRef] [PubMed]
- Sirey, T.M.; Roberts, K.; Haerty, W.; Bedoya-Reina, O.; Rogatti-Granados, S.; Tan, J.Y.; Li, N.; Heather, L.C.; Carter, R.N.; Cooper, S. The long non-coding RNA Cerox1 is a post transcriptional regulator of mitochondrial complex I catalytic activity. eLife 2019, 8, e45051. [Google Scholar] [CrossRef]
- Sun, X.; Wong, D. Long non-coding RNA-mediated regulation of glucose homeostasis and diabetes. Am. J. Cardiovasc. Dis. 2016, 6, 17–25. [Google Scholar]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 159. [Google Scholar] [CrossRef]
- Samarfard, S.; Ghorbani, A.; Karbanowicz, T.P.; Lim, Z.X.; Saedi, M.; Fariborzi, N.; McTaggart, A.R.; Izadpanah, K. Regulatory non-coding RNA: The core defense mechanism against plant pathogens. J. Biotechnol. 2022, 359, 82–94. [Google Scholar] [CrossRef]
- Xing, C.; Sun, S.-g.; Yue, Z.-Q.; Bai, F. Role of lncRNA LUCAT1 in cancer. Biomed. Pharmacother. 2021, 134, 111158. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Johnson, R. Global positioning system: Understanding long noncoding RNAs through subcellular localization. Mol. Cell 2019, 73, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Bridges, M.C.; Daulagala, A.C.; Kourtidis, A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021, 220, e202009045. [Google Scholar] [CrossRef]
- Kugel, J.F.; Goodrich, J.A. Non-coding RNAs: Key regulators of mammalian transcription. Trends Biochem. Sci. 2012, 37, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Melé, M.; Rinn, J.L. “Cat’s Cradling” the 3D genome by the act of LncRNA transcription. Mol. Cell 2016, 62, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Carninci, P. Long non-coding RNA modifies chromatin: Epigenetic silencing by long non-coding RNAs. Bioessays 2011, 33, 830–839. [Google Scholar] [CrossRef]
- Li, B.; Cai, L.; Liao, B.; Fu, X.; Bing, P.; Yang, J. Prediction of protein subcellular localization based on fusion of multi-view features. Molecules 2019, 24, 919. [Google Scholar] [CrossRef]
- Alaa, A.; Eldeib, A.M.; Metwally, A.A. Protein Subcellular Localization Prediction Based on Internal Micro-similarities of Markov Chains. In Proceedings of the 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Berlin, Germany, 23–27 July 2019; pp. 1355–1358. [Google Scholar]
- Gardy, J.L.; Brinkman, F.S. Methods for predicting bacterial protein subcellular localization. Nat. Rev. Microbiol. 2006, 4, 741–751. [Google Scholar] [CrossRef]
- Bhasin, M.; Garg, A.; Raghava, G.P.S. PSLpred: Prediction of subcellular localization of bacterial proteins. Bioinformatics 2005, 21, 2522–2524. [Google Scholar] [CrossRef]
- Gardy, J.L.; Spencer, C.; Wang, K.; Ester, M.; Tusnady, G.E.; Simon, I.; Hua, S.; DeFays, K.; Lambert, C.; Nakai, K. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 2003, 31, 3613–3617. [Google Scholar] [CrossRef]
- Hua, S.; Sun, Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics 2001, 17, 721–728. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Shen, Y.; Ding, Y.; Tang, J.; Zou, Q.; Guo, F. Critical evaluation of web-based prediction tools for human protein subcellular localization. Brief. Bioinform. 2020, 21, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Pan, X.; Yang, Y.; Huang, Y.; Shen, H.-B. The lncLocator: A subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 2018, 34, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-D.; Huang, Y.; Zhang, Z.-Y.; Zhao, Y.-W.; Wang, D.; Chen, W.; Chou, K.-C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Liang, Y.; Du, W.; Lv, W.; Li, Y. LncLocation: Efficient subcellular location prediction of long non-coding RNA-based multi-source heterogeneous feature fusion. Int. J. Mol. Sci. 2020, 21, 7271. [Google Scholar] [CrossRef]
- Gudenas, B.L.; Wang, L. Prediction of LncRNA subcellular localization with deep learning from sequence features. Sci. Rep. 2018, 8, 16385. [Google Scholar] [CrossRef]
- Zeng, M.; Wu, Y.; Lu, C.; Zhang, F.; Wu, F.-X.; Li, M. DeepLncLoc: A deep learning framework for long non-coding RNA subcellular localization prediction based on subsequence embedding. Brief. Bioinform. 2022, 23, bbab360. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, UK, 2013; Volume 26. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Spatial pyramid pooling in deep convolutional networks for visual recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 1904–1916. [Google Scholar] [CrossRef]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2017, 45, D135–D138. [Google Scholar] [CrossRef]
- Wen, X.; Gao, L.; Guo, X.; Li, X.; Huang, X.; Wang, Y.; Xu, H.; He, R.; Jia, C.; Liang, F. lncSLdb: A resource for long non-coding RNA subcellular localization. Database 2018, 2018, bay085. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jia, P.; Zhao, Z. Deep4mC: Systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Brief. Bioinform. 2021, 22, bbaa099. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, UK, 2017; Volume 30. [Google Scholar]
- Wang, D.; Zhang, Y.; Zhao, Y. LightGBM: An effective miRNA classification method in breast cancer patients. In Proceedings of the 2017 International Conference on Computational Biology and Bioinformatics, Newark, NJ, USA, 18–20 October 2017; pp. 7–11. [Google Scholar]
- Li, F.; Guo, X.; Jin, P.; Chen, J.; Xiang, D.; Song, J.; Coin, L.J.M. Porpoise: A new approach for accurate prediction of RNA pseudouridine sites. Brief. Bioinform. 2021, 22, bbab245. [Google Scholar] [CrossRef]
- Emami, N.; Ferdousi, R. AptaNet as a deep learning approach for aptamer–protein interaction prediction. Sci. Rep. 2021, 11, 6074. [Google Scholar] [CrossRef]
- Sperandei, S. Understanding logistic regression analysis. Biochem. Med. 2014, 24, 12–18. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Tyree, S.; Weinberger, K.Q.; Agrawal, K.; Paykin, J. Parallel boosted regression trees for web search ranking. In Proceedings of the 20th International Conference on World Wide Web, Hyderabad, India, 28 March–1 April 2011; pp. 387–396. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ridgeway, G. Generalized Boosted Models: A guide to the gbm package. Update 2007, 1, 2007. [Google Scholar]
- Song, Y.; Jiao, X.; Qiao, Y.; Liu, X.; Qiang, Y.; Liu, Z.; Zhang, L. Prediction of double-high biochemical indicators based on LightGBM and XGBoost. In Proceedings of the 2019 International Conference on Artificial Intelligence and Computer Science, Wuhan, China, 12–13 July 2019; pp. 189–193. [Google Scholar]
- Bi, Y.; Xiang, D.; Ge, Z.; Li, F.; Jia, C.; Song, J. An interpretable prediction model for identifying N7-methylguanosine sites based on XGBoost and SHAP. Mol. Ther.-Nucleic Acids 2020, 22, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Vo, T.-H.; Nguyen, Q.H.; Do, T.T.; Nguyen, T.-N.; Rahardja, S.; Nguyen, B.P. iPseU-NCP: Identifying RNA pseudouridine sites using random forest and NCP-encoded features. BMC Genom. 2019, 20, 971. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V.; Tang, Y.; Cho, H.; Chen, K. Xgboost: Extreme gradient boosting. R Package Version 0.4-2 2015, 1, 1–4. [Google Scholar]
- Hearst, M.A.; Dumais, S.T.; Osuna, E.; Platt, J.; Scholkopf, B. Support vector machines. IEEE Intell. Syst. Appl. 1998, 13, 18–28. [Google Scholar] [CrossRef]
- Pal, M. Random forest classifier for remote sensing classification. Int. J. Remote Sens. 2005, 26, 217–222. [Google Scholar] [CrossRef]
- Wright, R.E. Logistic regression. In Reading and Understanding Multivariate Statistics; American Psychological Association: Washington, DC, USA, 1995; pp. 217–244. [Google Scholar]
- Ruck, D.W.; Rogers, S.K.; Kabrisky, M. Feature selection using a multilayer perceptron. J. Neural Netw. Comput. 1990, 2, 40–48. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
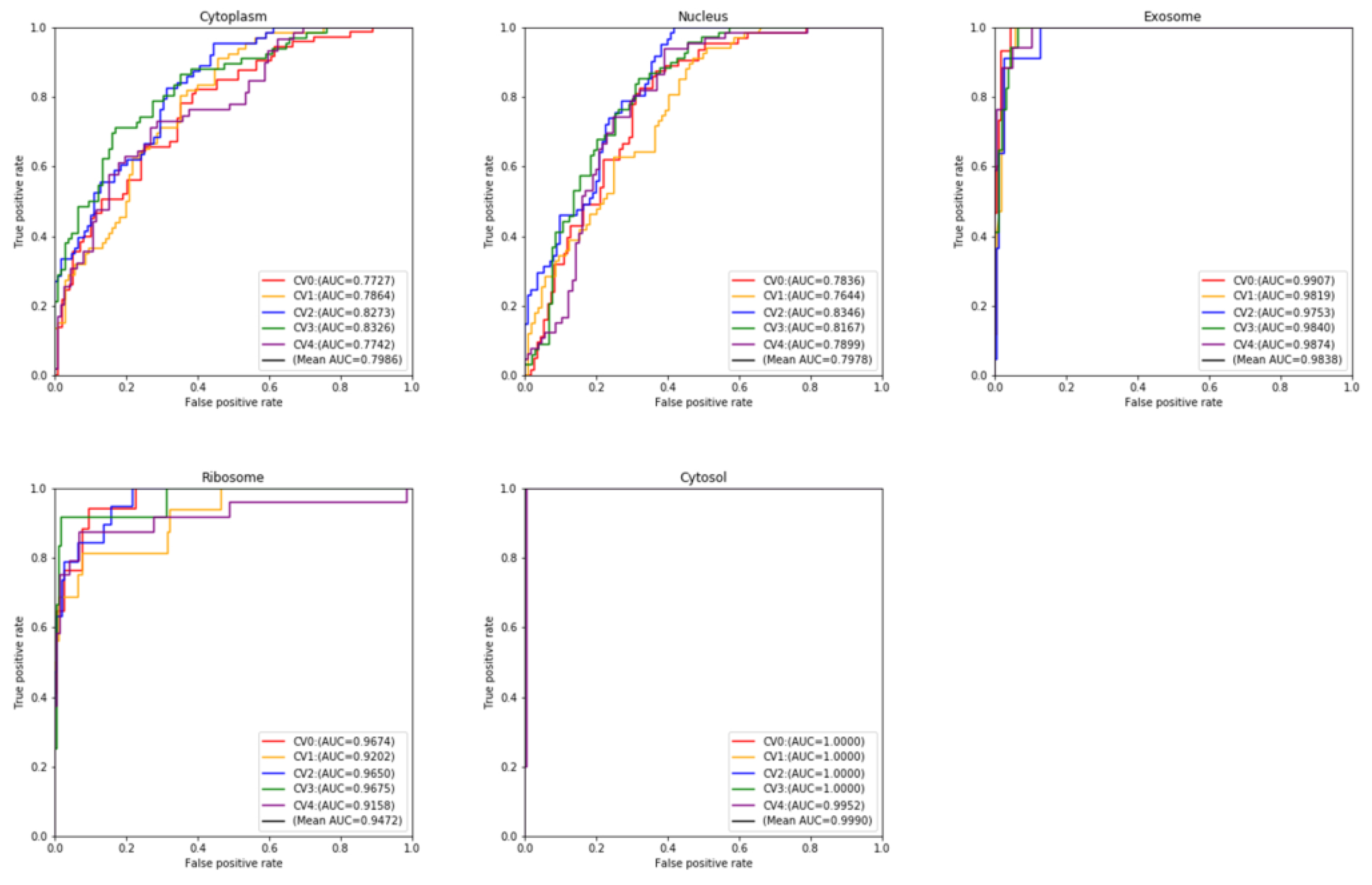{kind=link}
| Feature Type | ACC | AUC | Macro F-Measure |
|---|---|---|---|
| PSTNPSSMC | 0.673 | 0.895 | 0.776 |
| RCKmer | 0.512 | 0.616 | 0.290 |
| CKSNAP | 0.467 | 0.592 | 0.267 |
| NAC | 0.466 | 0.590 | 0.240 |
| ENAC | 0.463 | 0.612 | 0.261 |
| NCP | 0.454 | 0.591 | 0.246 |
| ANF | 0.429 | 0.531 | 0.233 |
| Feature Type | ACC | AUC | Macro F-Measure |
|---|---|---|---|
| PSTNPSSMC | 0.673 | 0.895 | 0.776 |
| PSTNPSSMC + RCKmer | 0.696 | 0.904 | 0.772 |
| PSTNPSSMC + CKSNAP | 0.687 | 0.899 | 0.758 |
| PSTNPSSMC + RCKmer + CKSNAP | 0.695 | 0.906 | 0.775 |
| PSTNPSSMC + RCKmer + NAC | 0.686 | 0.901 | 0.769 |
| PSTNPSSMC + CKSNAP + NAC | 0.678 | 0.905 | 0.780 |
| Methods | ACC | AUC | Macro F-Measure |
|---|---|---|---|
| LightGBM | 0.673 | 0.895 | 0.776 |
| XGBoost | 0.645 | 0.895 | 0.735 |
| SVM | 0.602 | 0.882 | 0.700 |
| Random Forest | 0.632 | 0.890 | 0.752 |
| Logical regression | 0.395 | 0.839 | 0.242 |
| Multilayer perceptron | 0.557 | 0.854 | 0.664 |
| k | ACC | AUC | Macro F-Measure |
|---|---|---|---|
| 2 | 0.692 | 0.906 | 0.785 |
| 3 | 0.696 | 0.904 | 0.772 |
| 4 | 0.698 | 0.903 | 0.793 |
| 5 | 0.700 | 0.905 | 0.779 |
| 6 | 0.672 | 0.897 | 0.759 |
| Location | ACC | AUC | Macro F-Measure |
|---|---|---|---|
| The first 166 of lncRNAs | 0.703 | 0.904 | 0.792 |
| The last 166 of lncRNAs | 0.607 | 0.887 | 0.704 |
| Random 166 of lncRNAs | 0.583 | 0.862 | 0.671 |
| Predictor | ACC | Macro F-Measure | AUC |
|---|---|---|---|
| DeepLncLoc | 0.548 | 0.420 | 0.820 |
| LightGBM-LncLoc | 0.706 | 0.786 | 0.904 |
| Predictor | Macro Precision | Macro Recall | Macro F-Measure | ACC |
|---|---|---|---|---|
| iLoc-lncRNA | 0.488 | 0.445 | 0.458 | 0.507 |
| DeepLncLoc | 0.702 | 0.524 | 0.563 | 0.537 |
| LightGBM-LncLoc | 0.779 | 0.525 | 0.576 | 0.567 |
| Predictor | LightGBM-LncLoc | DeepLncLoc | iLoc-lncRNA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Precision | Recall | F1 | Precision | Recall | F1 | Precision | Recall | F1 | |
| Cytoplasm | 0.667 | 0.400 | 0.500 | 0.778 | 0.350 | 0.483 | 0.553 | 0.700 | 0.618 |
| Nucleus | 0.429 | 0.900 | 0.581 | 0.400 | 0.800 | 0.533 | 0.467 | 0.350 | 0.400 |
| Ribosome | 0.800 | 0.400 | 0.533 | 0.500 | 0.400 | 0.444 | 0.333 | 0.500 | 0.316 |
| Cytosol | 1.000 | 0.500 | 0.667 | 0.833 | 0.500 | 0.625 | null | null | null |
| Exosome | 1.000 | 0.429 | 0.600 | 1.000 | 0.571 | 0.727 | 0.600 | 0.429 | 0.500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, J.; Zheng, P.; Qi, Y.; Huang, G. LightGBM-LncLoc: A LightGBM-Based Computational Predictor for Recognizing Long Non-Coding RNA Subcellular Localization. Mathematics 2023, 11, 602. https://doi.org/10.3390/math11030602
Lyu J, Zheng P, Qi Y, Huang G. LightGBM-LncLoc: A LightGBM-Based Computational Predictor for Recognizing Long Non-Coding RNA Subcellular Localization. Mathematics. 2023; 11(3):602. https://doi.org/10.3390/math11030602
Chicago/Turabian StyleLyu, Jianyi, Peijie Zheng, Yue Qi, and Guohua Huang. 2023. "LightGBM-LncLoc: A LightGBM-Based Computational Predictor for Recognizing Long Non-Coding RNA Subcellular Localization" Mathematics 11, no. 3: 602. https://doi.org/10.3390/math11030602
APA StyleLyu, J., Zheng, P., Qi, Y., & Huang, G. (2023). LightGBM-LncLoc: A LightGBM-Based Computational Predictor for Recognizing Long Non-Coding RNA Subcellular Localization. Mathematics, 11(3), 602. https://doi.org/10.3390/math11030602