
COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation

 , ,
, ,  , ,
, ,  ,
,
Abstract
:1. Introduction
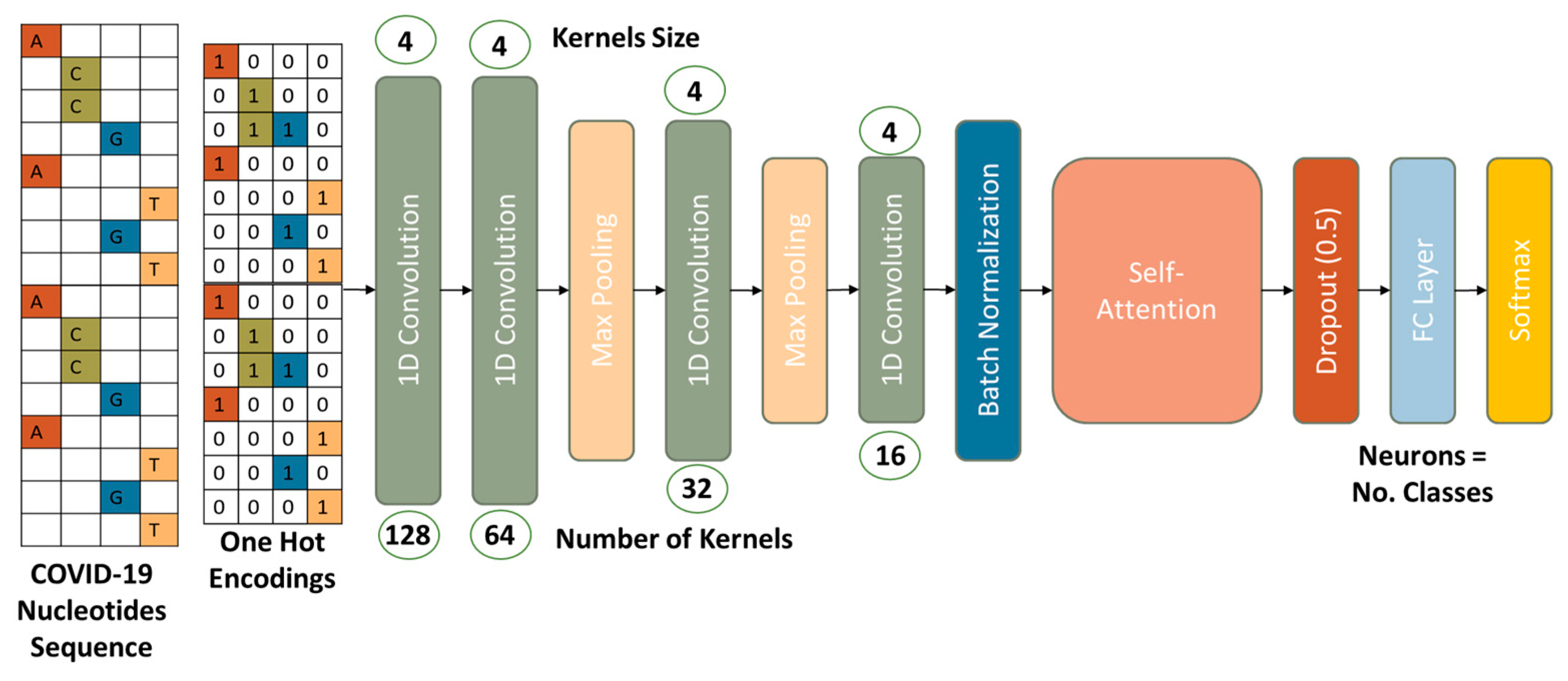
- We proposed a convolutional neural network (CNN) to classify the COVID-19 variants. The proposed model utilizes 1D convolutions, batch normalization, and self-attention layers to extract discriminative features from the nucleotide sequences. Self-attention is employed to cope with the relationship and mutation of adenine (A), cytosine (C), guanine (G), and thymine (T) in the sequence.
- The detection of new variants of COVID-19 is an important task for the scientific community to develop better healthcare and economic plans to deal with its spread. To the best of our knowledge, our proposed framework is the first attempt to detect new variants. Our framework employs uncertainty calculation via entropy followed by an optimum threshold to detect unknown variants of COVID-19.
- We introduce a novel COVID-19 new variant generation technique, which is used to evaluate the proposed variant detection technique. Our variational autoencoder-decoder (VAE) network is able to predict new sequences with significant similarity with the original coronavirus and its variants using the BLAST method. We also believe that the proactive nucleotide sequence prediction of possible new variants of COVID-19 could assist vaccine providers in increasing the efficacy of vaccines.
2. Related Works
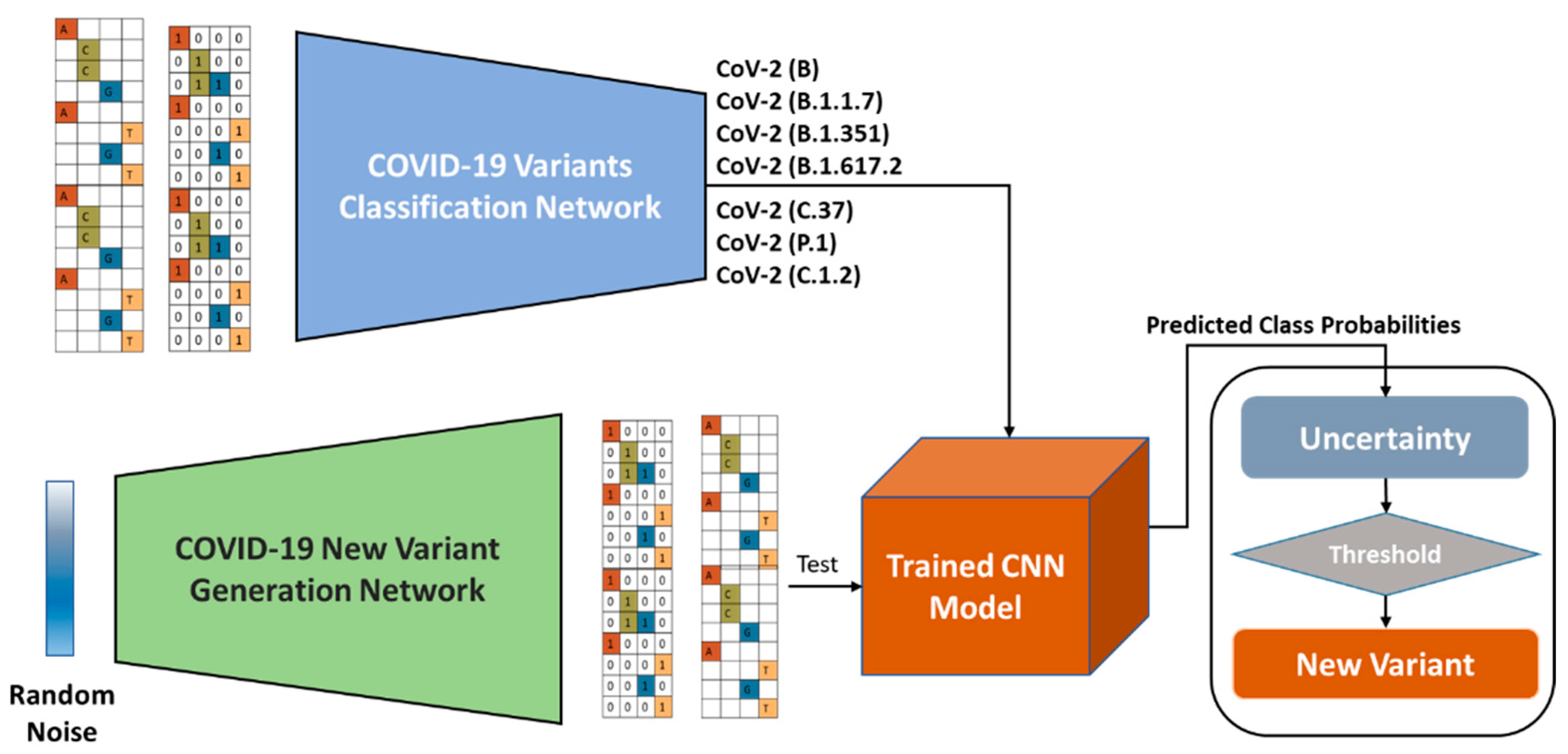
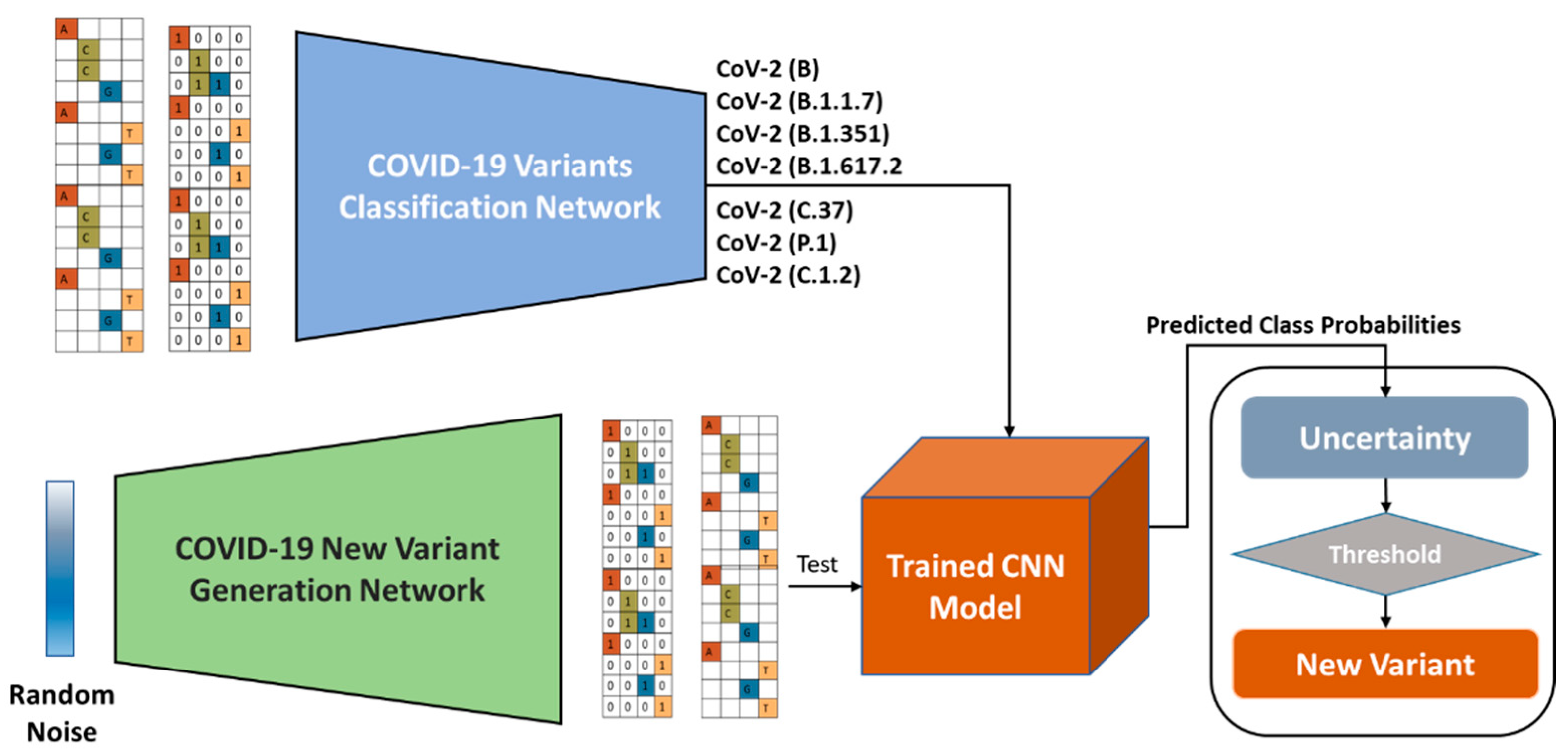
3. COVID-19 Nucleotide Sequences Analysis Framework
3.1. Nucleotides Data Preprocessing
3.2. COVID-19 Variants Classification Network
3.3. Self-Attention
3.4. New Variant Detection
3.5. COVID-19 Variant Generation
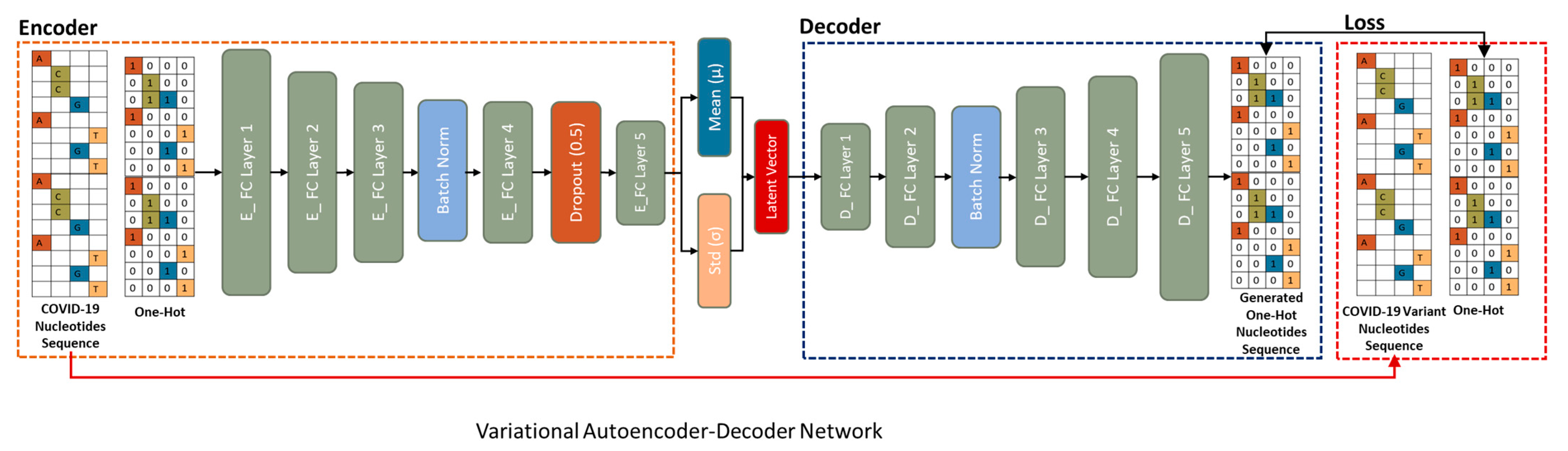
3.6. Variational Autoencoder-Decoder
| Algorithm 1: COVID-19 variant prediction |
| Steps: 1. M ← Load Attention-CNN model 2. seq ← Load nucleotide sequences 3. th ← 0.15 # threshold for uncertainty prediction 4. one_seq ← ONE_HOT_Conversion (seq) 5. preds ← M (one_seq) 6. score ← ENTROPY (preds) 7. if (score > th): print (‘New variant’) else: varinat ← MAX (preds) print (varinat) |
4. Experimental Results and Discussion
4.1. Dataset
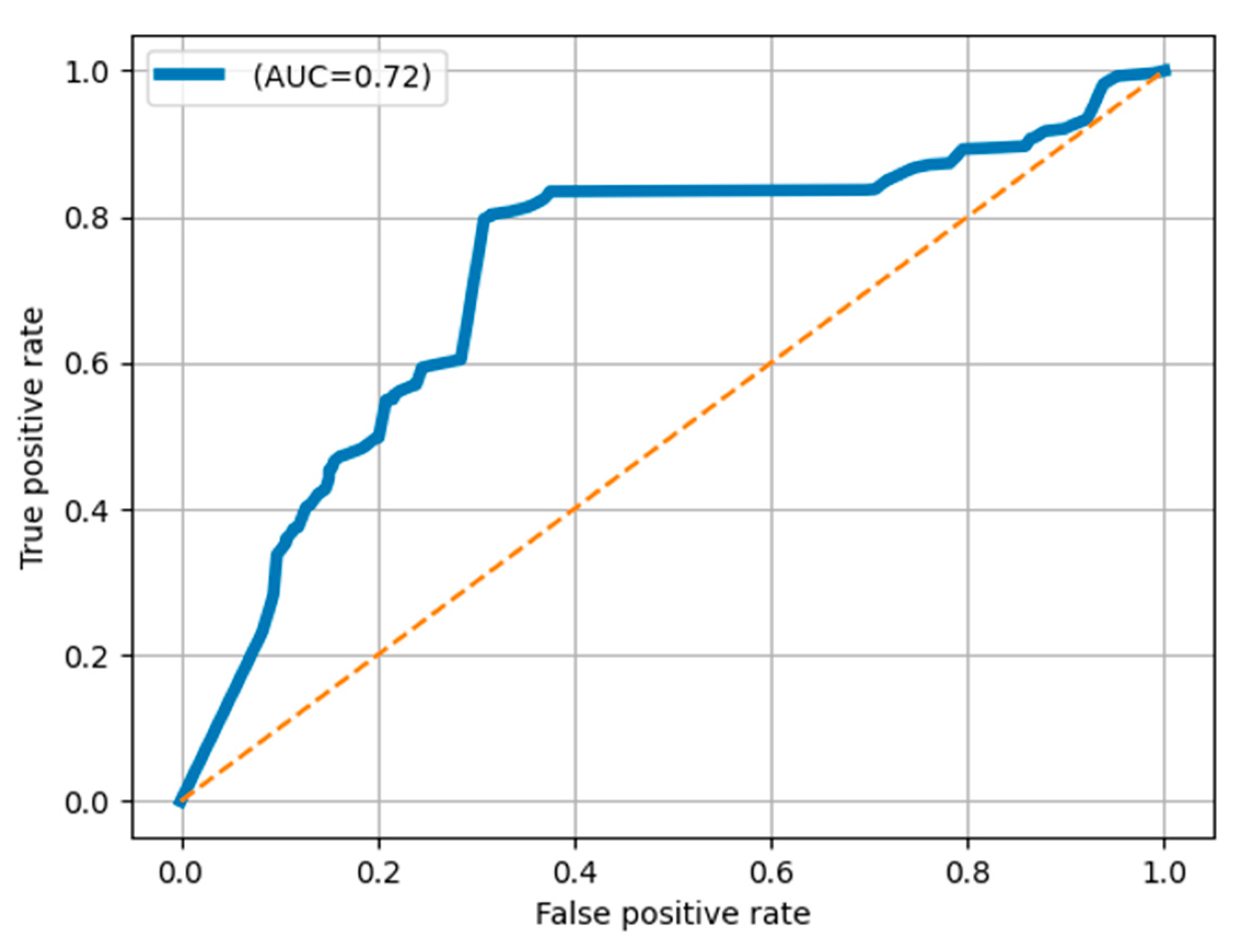
4.2. Evaluation of the Classification Network
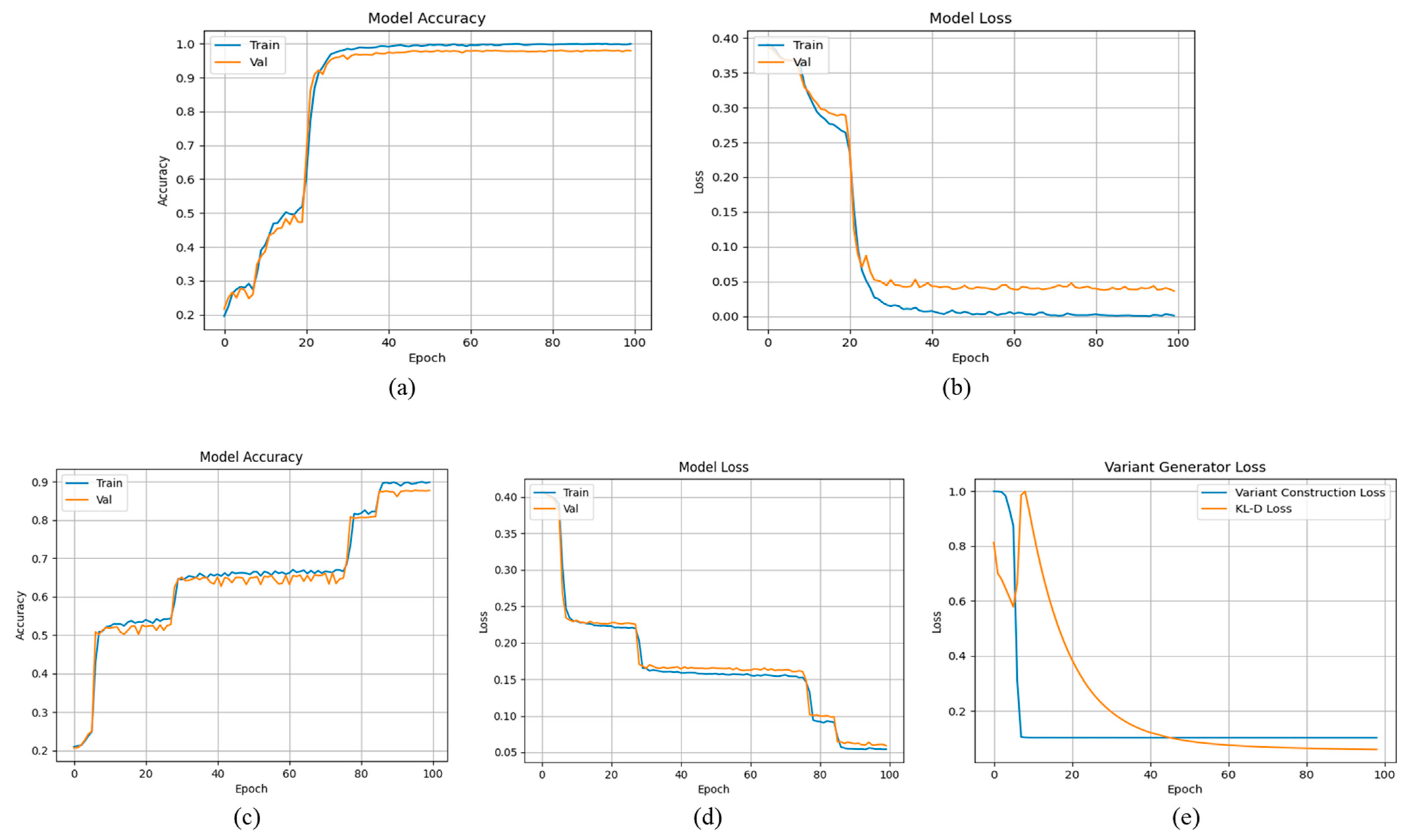
4.3. Evaluation of the New Variant Generation Network
4.4. New Variant Prediction Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lv, M.; Luo, X.; Estill, J.; Liu, Y.; Ren, M.; Wang, J.; Wang, Q.; Zhao, S.; Wang, X.; Yang, S.J.E. Coronavirus disease (COVID-19): A scoping review. Eurosurveillance 2020, 25, 2000125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. COVID-19 Weekly Epidemiological Update, 54th ed.; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Abdulkareem, K.H.; Mohammed, M.A.; Salim, A.; Arif, M.; Geman, O.; Gupta, D.; Khanna, A. Realizing an effective COVID-19 diagnosis system based on machine learning and IOT in smart hospital environment. IEEE Internet Things J. 2021, 8, 15919–15928. [Google Scholar] [CrossRef]
- Esbin, M.N.; Whitney, O.N.; Chong, S.; Maurer, A.; Darzacq, X.; Tjian, R. Overcoming the bottleneck to widespread testing: A rapid review of nucleic acid testing approaches for COVID-19 detection. RNA 2020, 26, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Delgado, E.J.; Cabezas, X.; Martin-Barreiro, C.; Leiva, V.; Rojas, F. An Equity-Based Optimization Model to Solve the Location Problem for Healthcare Centers Applied to Hospital Beds and COVID-19 Vaccination. Mathematics 2022, 10, 1825. [Google Scholar] [CrossRef]
- Akram, T.; Attique, M.; Gul, S.; Shahzad, A.; Altaf, M.; Naqvi, S.S.R.; Damaševičius, R.; Maskeliūnas, R. A novel framework for rapid diagnosis of COVID-19 on computed tomography scans. Pattern Anal. Appl. 2021, 24, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Sahlol, A.T.; Yousri, D.; Ewees, A.A.; Al-Qaness, M.A.; Damasevicius, R.; Abd Elaziz, M. COVID-19 image classification using deep features and fractional-order marine predators algorithm. Sci. Rep. 2020, 10, 15364. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, H. Machine Learning Methods for COVID-19 Prediction Using Human Genomic Data. Proceedings 2021, 74, 20. [Google Scholar] [CrossRef]
- Arslan, H.; Arslan, H. A new COVID-19 detection method from human genome sequences using CpG island features and KNN classifier. Eng. Sci. Technol. Int. J. 2021, 24, 839–847. [Google Scholar] [CrossRef]
- Cortés-Carvajal, P.D.; Cubilla-Montilla, M.; González-Cortés, D.R. Estimation of the instantaneous reproduction number and its confidence interval for modeling the COVID-19 pandemic. Mathematics 2022, 10, 287. [Google Scholar] [CrossRef]
- Sharma, N.; Krishnan, P.; Kumar, R.; Ramoji, S.; Chetupalli, S.R.; Ghosh, P.K.; Ganapathy, S. Coswara—A Database of Breathing, Cough, and Voice Sounds for COVID-19 Diagnosis. arXiv 2020, arXiv:2005.10548. [Google Scholar]
- Asraf, A.; Islam, M.Z.; Haque, M.R.; Islam, M.M. Deep learning applications to combat novel coronavirus (COVID-19) pandemic. SN Comput. Sci. 2020, 1, 363. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gao, B.; Sabnis, R.; Sun, Q. Nucleic Transformer: Deep Learning on Nucleic Acids with Self-Attention and Convolutions. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dasari, C.M.; Bhukya, R. Explainable deep neural networks for novel viral genome prediction. Appl. Intell. 2022, 52, 3002–3017. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kang, B.; Ma, J.; Zeng, X.; Xiao, M.; Guo, J.; Cai, M.; Yang, J.; Li, Y.; Meng, X.; et al. A deep learning algorithm using CT images to screen for Corona Virus Disease (COVID-19). Eur. Radiol. 2021, 31, 6096–6104. [Google Scholar] [CrossRef]
- Barstugan, M.; Ozkaya, U.; Ozturk, S. Coronavirus (COVID-19) classification using ct images by machine learning methods. arXiv 2020, arXiv:2003.09424. [Google Scholar]
- Gozes, O.; Frid-Adar, M.; Greenspan, H.; Browning, P.D.; Zhang, H.; Ji, W.; Bernheim, A.; Siegel, E. Rapid ai development cycle for the coronavirus (COVID-19) pandemic: Initial results for automated detection & patient monitoring using deep learning ct image analysis. arXiv 2020, arXiv:2003.05037. [Google Scholar]
- Özkaya, U.; Öztürk, Ş.; Barstugan, M. Coronavirus (COVID-19) classification using deep features fusion and ranking technique. In Big Data Analytics and Artificial Intelligence against COVID-19: Innovation Vision and Approach; Springer: Berlin/Heidelberg, Germany, 2020; pp. 281–295. [Google Scholar]
- Muhammad, L.; Algehyne, E.A.; Usman, S.S.; Ahmad, A.; Chakraborty, C.; Mohammed, I.A. Supervised machine learning models for prediction of COVID-19 infection using epidemiology dataset. SN Comput. Sci. 2021, 2, 11. [Google Scholar] [CrossRef]
- Narin, A.; Kaya, C.; Pamuk, Z. Automatic detection of coronavirus disease (COVID-19) using X-ray images and deep convolutional neural networks. Pattern Anal. Appl. 2021, 24, 1207–1220. [Google Scholar] [CrossRef]
- Song, Y.; Zheng, S.; Li, L.; Zhang, X.; Zhang, X.; Huang, Z.; Chen, J.; Wang, R.; Zhao, H.; Zha, Y.; et al. Deep learning enables accurate diagnosis of novel coronavirus (COVID-19) with CT images. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 2775–2780. [Google Scholar] [CrossRef]
- Pan, X.; Rijnbeek, P.; Yan, J.; Shen, H.-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genom. 2018, 19, 511. [Google Scholar] [CrossRef] [Green Version]
- Ullah, A.; Muhammad, K.; Hussain, T.; Baik, S.W.; De Albuquerque, V.H.C. Event-oriented 3D convolutional features selection and hash codes generation using PCA for video retrieval. IEEE Access 2020, 8, 196529–196540. [Google Scholar] [CrossRef]
- Muhammad, K.; Ullah, A.; Imran, A.S.; Sajjad, M.; Kiran, M.S.; Sannino, G.; de Albuquerque, V.H.C. Human action recognition using attention based LSTM network with dilated CNN features. Future Gener. Comput. Syst. 2021, 125, 820–830. [Google Scholar] [CrossRef]
- Song, Y.; Fu, Q.; Wang, Y.-F.; Wang, X. Divergence-based cross entropy and uncertainty measures of Atanassov’s intuitionistic fuzzy sets with their application in decision making. Appl. Soft Comput. 2019, 84, 105703. [Google Scholar] [CrossRef]
- Killoran, N.; Lee, L.J.; Delong, A.; Duvenaud, D.; Frey, B.J. Generating and designing DNA with deep generative models. arXiv 2017, arXiv:1712.06148. [Google Scholar]
- Rangasamy, M.; Chesneau, C.; Martin-Barreiro, C.; Leiva, V. On a Novel Dynamics of SEIR Epidemic Models with a Potential Application to COVID-19. Symmetry 2022, 14, 1436. [Google Scholar] [CrossRef]
- Pu, Y.; Gan, Z.; Henao, R.; Yuan, X.; Li, C.; Stevens, A.; Carin, L. Variational autoencoder for deep learning of images, labels and captions. Adv. Neural Inf. Process. Syst. 2016, 29, 2352–2360. [Google Scholar]
- Ullah, W.; Muhammad, K.; Ul Haq, I.; Ullah, A.; Ullah Khattak, S.; Sajjad, M. Splicing sites prediction of human genome using machine learning techniques. Multimed. Tools Appl. 2021, 80, 30439–30460. [Google Scholar] [CrossRef]
- Hassanzadeh, H.R.; Wang, M.D. DeeperBind: Enhancing prediction of sequence specificities of DNA binding proteins. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 178–183. [Google Scholar]
- Khan, S.U.; Baik, R. MPPIF-Net: Identification of Plasmodium Falciparum Parasite Mitochondrial Proteins Using Deep Features with Multilayer Bi-directional LSTM. Processes 2020, 8, 725. [Google Scholar] [CrossRef]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- Likic, V. The Needleman-Wunsch Algorithm for Sequence Alignment; Lecture given at the 7th Melbourne Bioinformatics Course; Bi021 Molecular Science and Biotechnology Institute, University of Melbourne: Melbourne, Australia, 2008; pp. 1–46. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
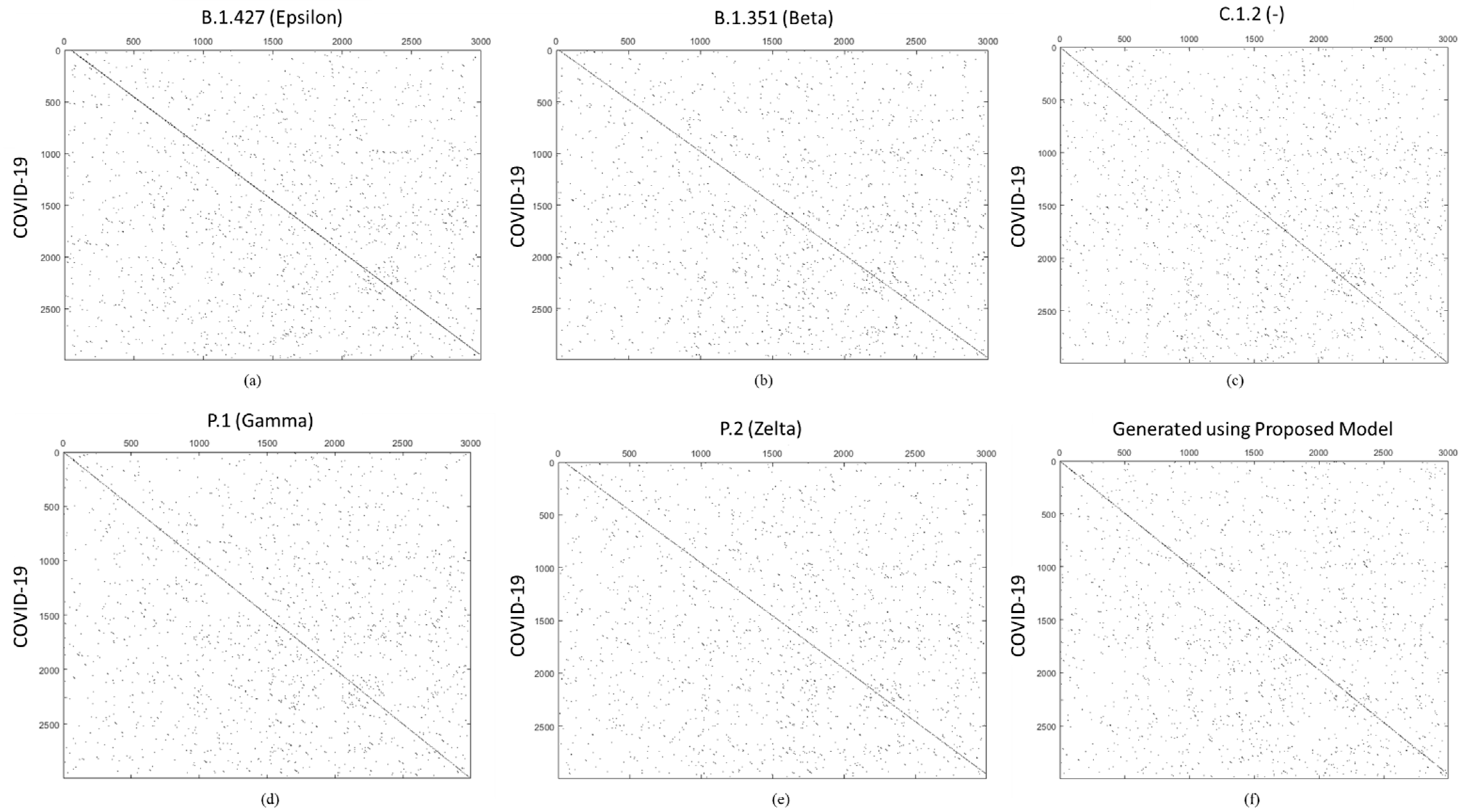{kind=link}
{kind=link}
{kind=link}
| No. | Variants Scientific Codes | Known Names | No. of Nucleotide Sequences Length = 30,000 | No. of Protein Sequences Length = 3000 | Classification Network Training | New Variant Prediction Test |
|---|---|---|---|---|---|---|
| 1 | B | SARS-CoV-2 | 1500 | 1109 | 🗸 | - |
| 2 | B.1.1.7 | CoV-Alpha | 1500 | 1053 | 🗸 | - |
| 3 | B.1.351 | CoV-Beta | 1500 | 1058 | 🗸 | - |
| 4 | B.1.617.2 | CoV-Dalta | 1015 | 1078 | 🗸 | - |
| 5 | C.37 | CoV-Lambda | 661 | 1056 | 🗸 | - |
| 6 | P.1 | CoV-Gamma | 1500 | 1073 | 🗸 | - |
| 7 | B.1.525 | CoV-Eta | 1500 | 1720 | 🗸 | - |
| 8 | C.1.2 | - | 119 | 14 | - | 🗸 |
| 9 | P.2 | CoV-Zeta | 1047 | 1059 | - | 🗸 |
| 10 | B.1.427 | CoV- Epsilon | 1500 | 1071 | - | 🗸 |
| Method | Overall Accuracy |
|---|---|
| DNC [30] | 64.7 |
| TNC [30] | 63.1 |
| TetraNC [30] | 64.5 |
| Composite NC [30] | 66.9 |
| DeepBind [31] | 85.6 |
| DeeperBind [31] | 89.2 |
| MPPIF-Net [32] | 85.7 |
| Our baseline (Protein) | 88.6 |
| Our Attention-CNN (Protein) | 90.6 |
| Our baseline (Nucleotides) | 93.4 |
| Our Attention-CNN (Nucleotides) | 96.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, A.; Malik, K.M.; Saudagar, A.K.J.; Khan, M.B.; Hasanat, M.H.A.; AlTameem, A.; AlKhathami, M.; Sajjad, M. COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation. Mathematics 2022, 10, 4267. https://doi.org/10.3390/math10224267
Ullah A, Malik KM, Saudagar AKJ, Khan MB, Hasanat MHA, AlTameem A, AlKhathami M, Sajjad M. COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation. Mathematics. 2022; 10(22):4267. https://doi.org/10.3390/math10224267
Chicago/Turabian StyleUllah, Amin, Khalid Mahmood Malik, Abdul Khader Jilani Saudagar, Muhammad Badruddin Khan, Mozaherul Hoque Abul Hasanat, Abdullah AlTameem, Mohammed AlKhathami, and Muhammad Sajjad. 2022. "COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation" Mathematics 10, no. 22: 4267. https://doi.org/10.3390/math10224267
APA StyleUllah, A., Malik, K. M., Saudagar, A. K. J., Khan, M. B., Hasanat, M. H. A., AlTameem, A., AlKhathami, M., & Sajjad, M. (2022). COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation. Mathematics, 10(22), 4267. https://doi.org/10.3390/math10224267