
Mathematical Model of Pancreatic Cancer Cell Dynamics Considering the Set of Sequential Mutations and Interaction with the Immune System

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
- The process of formation of precancerous and cancerous cells represents a successive chain of mutations over a relatively long period of time (10 to 20 years). During this time, the disease often causes very limited noticeable symptoms, and the cancer has often already spread at the time of diagnosis, with the most common sites of metastasis being the liver, lung, and peritoneum [3].
Framework for Quasispecies Dynamics
- (a)
- is a smooth function ;
- (b)
- ;
- (c)
- If , then ;
- (d)
- Function has only one maximum at the point , .
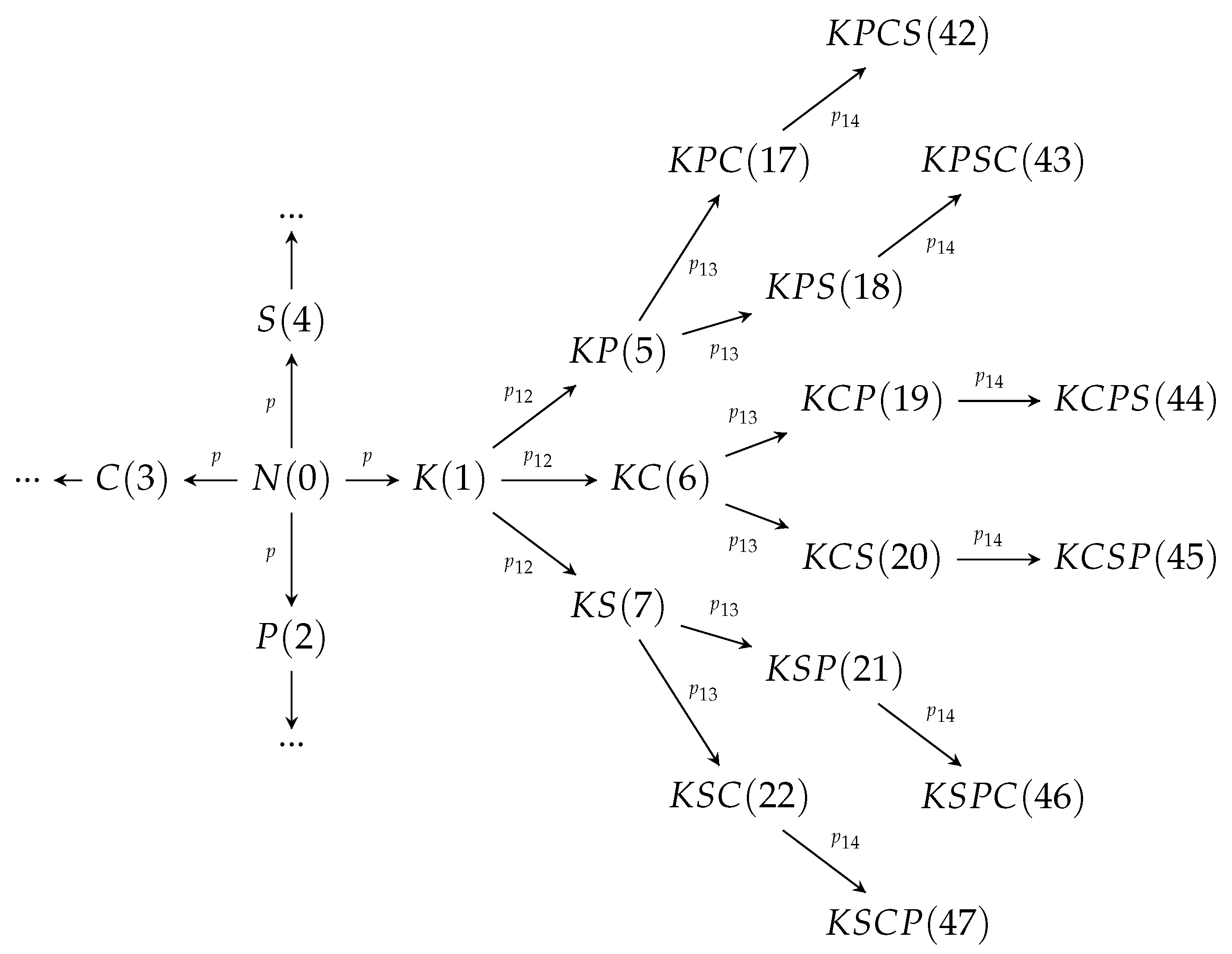
2. Development of the Cancer Mutations
3. Model Calibration for Cancer and Precancerous Cell Dynamics
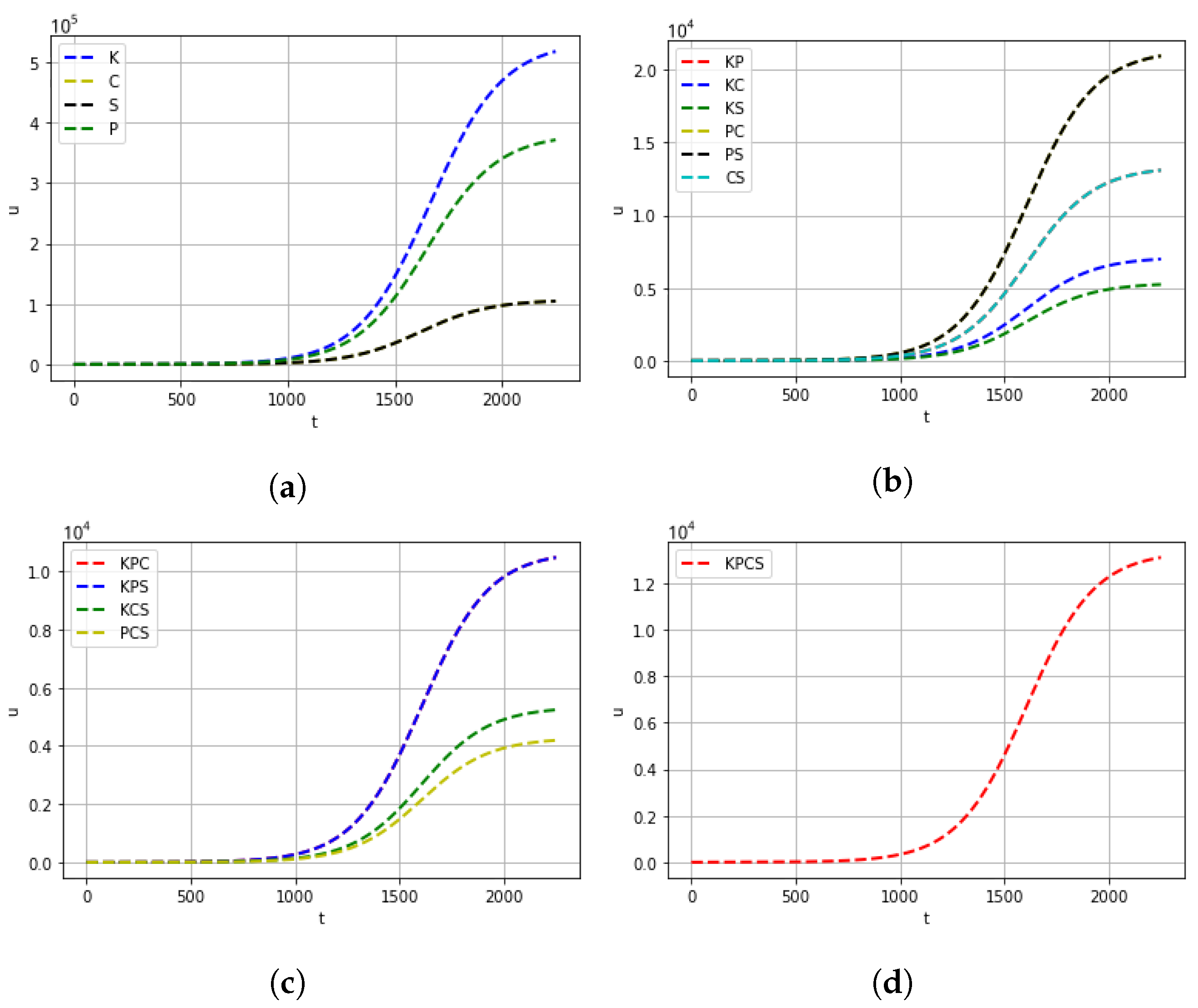
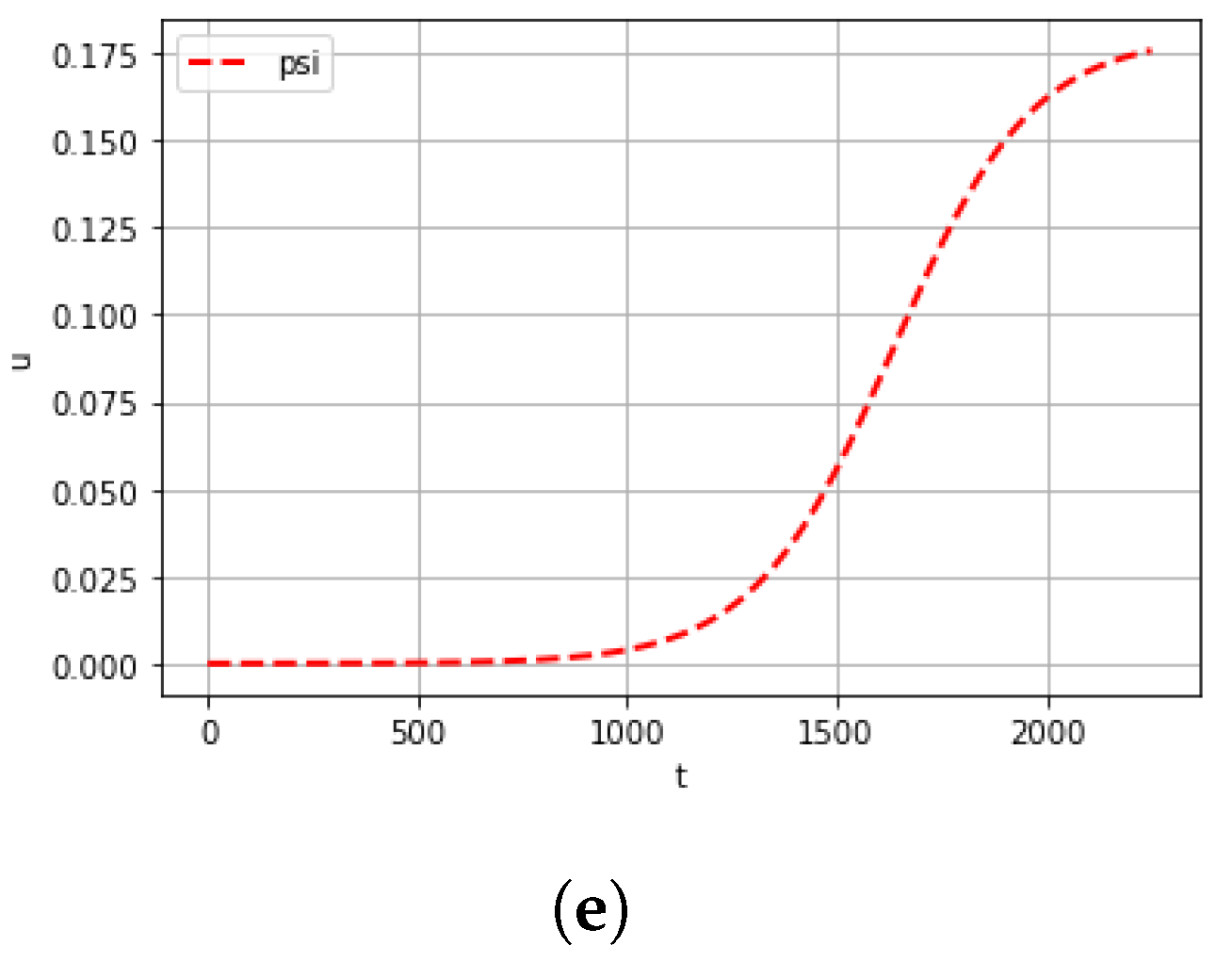
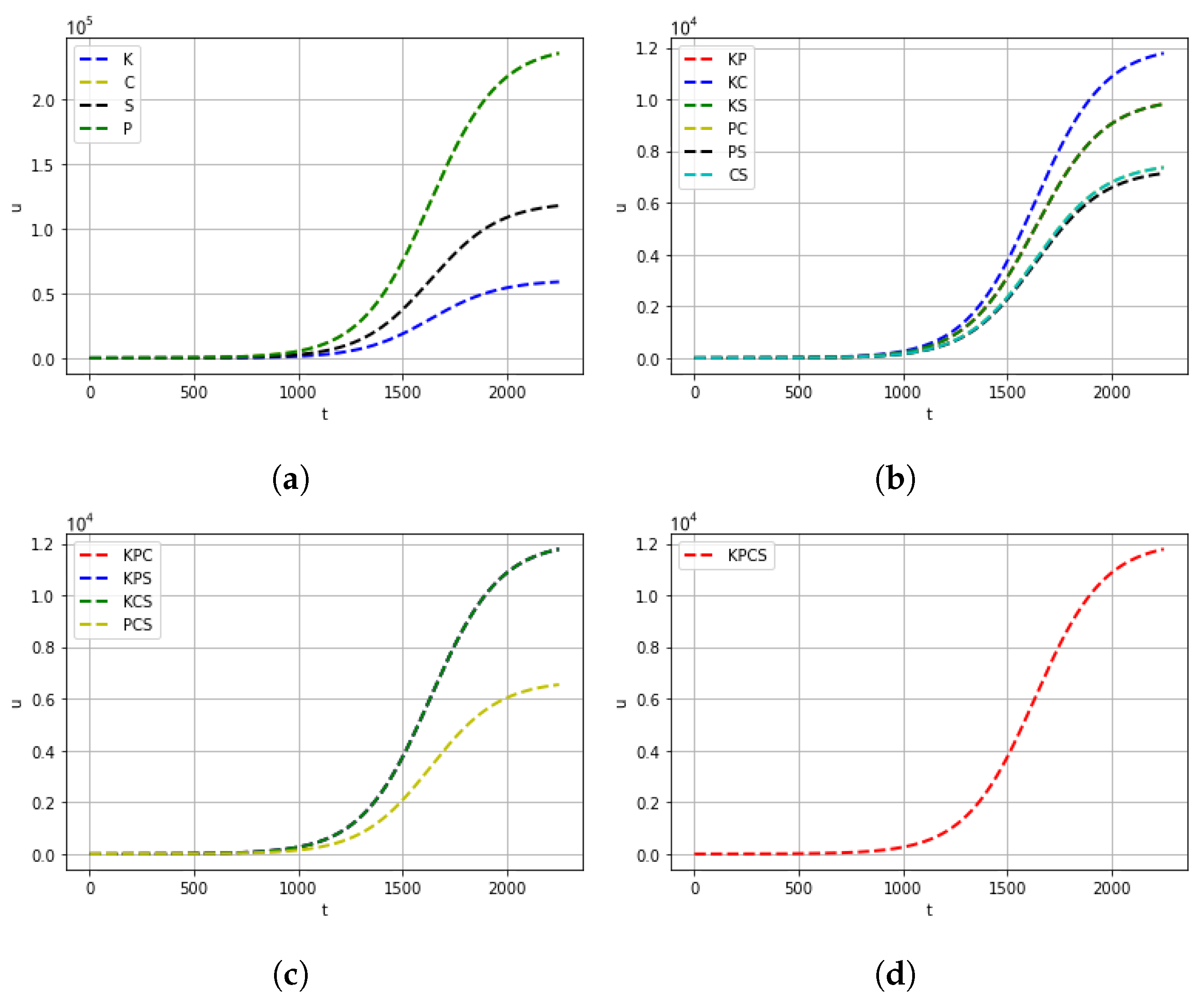
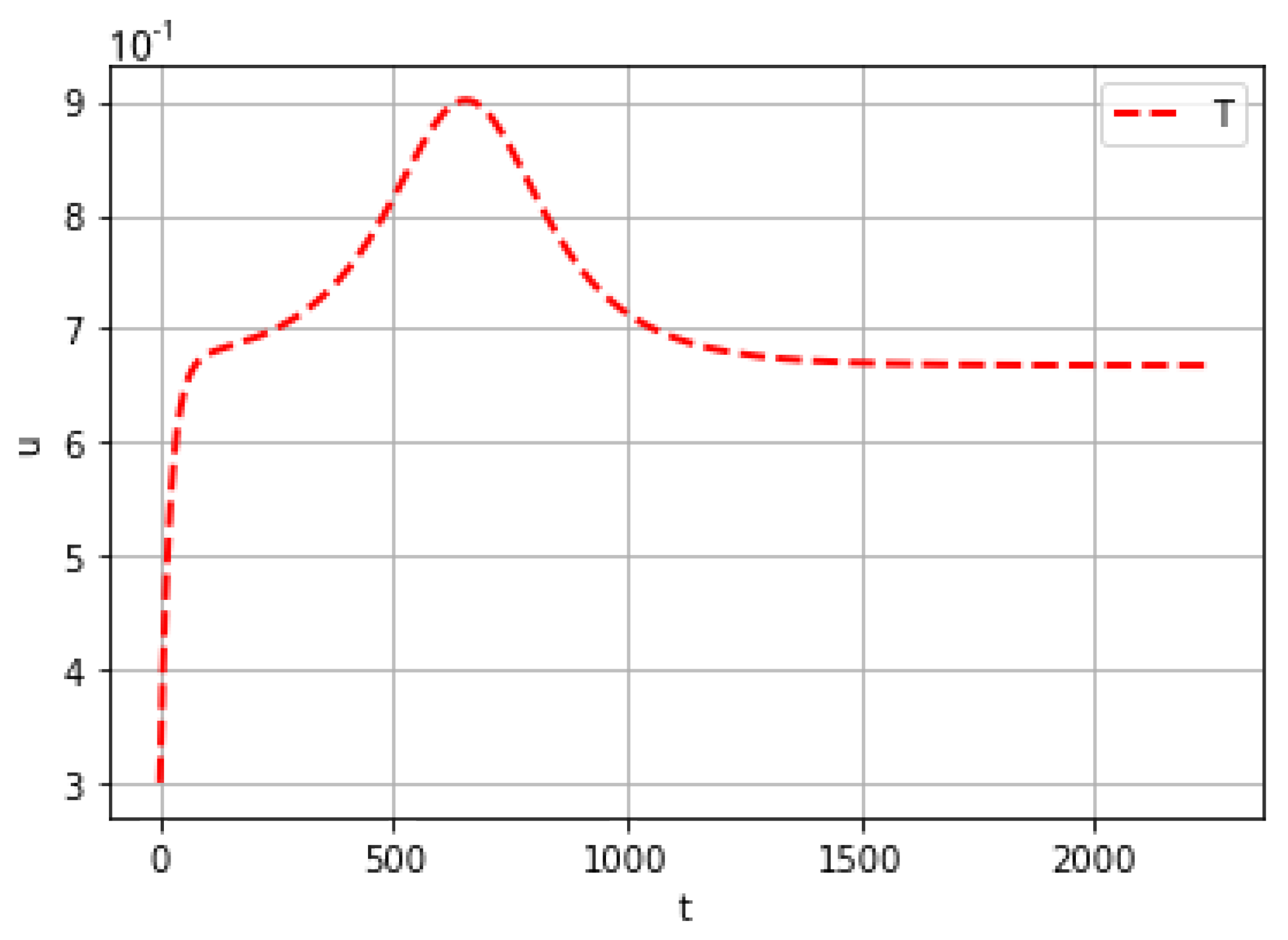
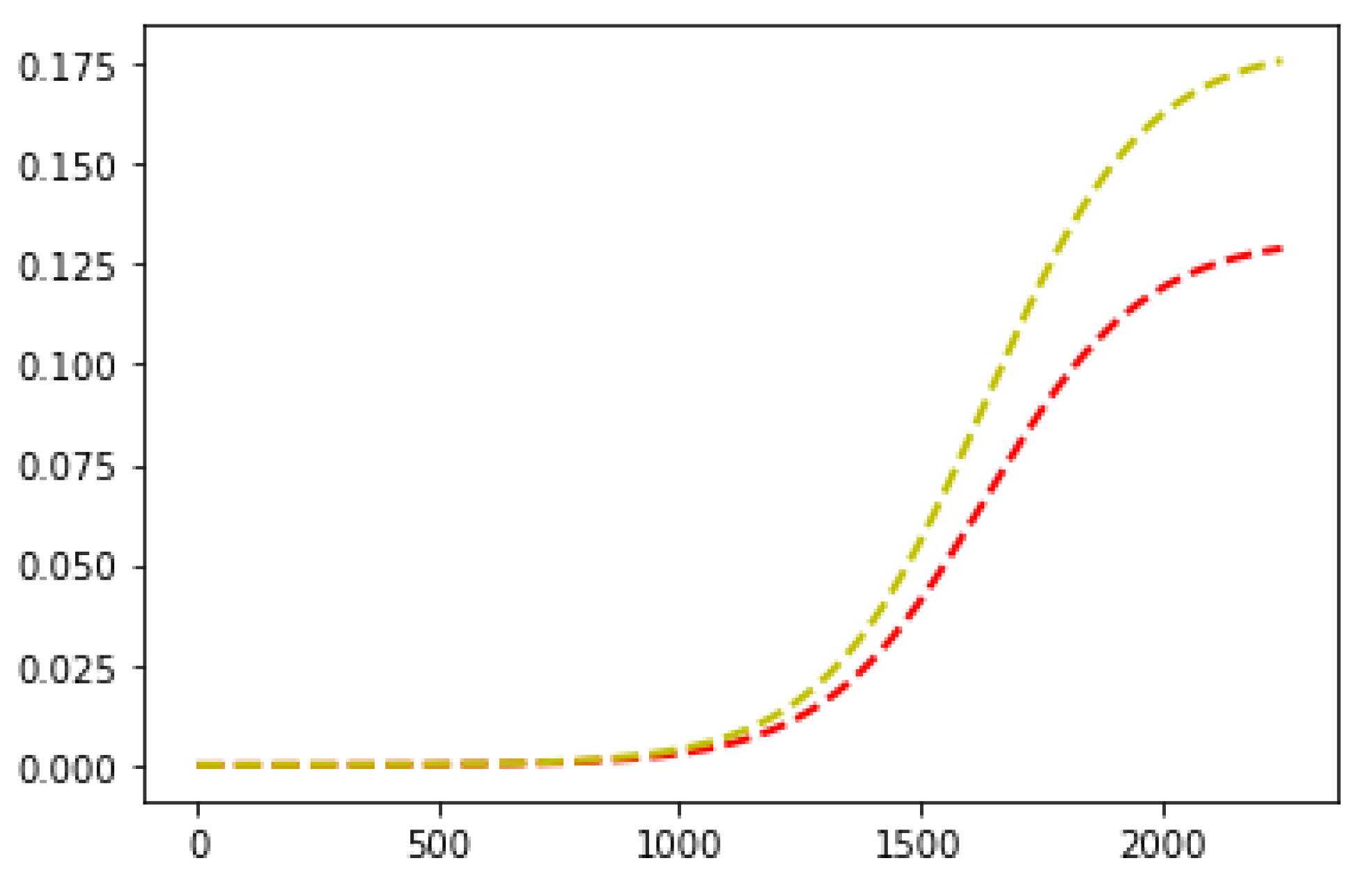
4. Computational Experiments
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| CTL | effector CD8+ T lymphocyte |
| ODE | ordinary differential equation |
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Lu, J.; Yu, R.; Liu, R.; Liang, X.; Sun, J.; Zhang, H.; Wu, H.; Zhang, Z.; Shao, Y.W.; Guo, J.; et al. Genetic aberrations in Chinese pancreatic cancer patients and their association with anatomic location and disease outcomes. Cancer Med. 2021, 10, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xu, J.W.; Cheng, Y.G.; Gao, J.Y.; Hu, S.Y.; Wang, L.; Zhan, H.X. Early detection of pancreatic cancer: Where are we now and where are we going?: Early detection of pancreatic cancer. Int. J. Cancer 2017, 141, 231–241. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Louzoun, Y.; Xue, C.; Lesinski, G.B.; Friedman, A. A mathematical model for pancreatic cancer growth and treatments. J. Theor. Biol. 2014, 351, 74–82. [Google Scholar] [CrossRef]
- Gaspar, N.J.; Li, L.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of Transforming Growth Factor β Signaling Reduces Pancreatic Adenocarcinoma Growth and Invasiveness. Mol. Pharmacol. 2007, 72, 152–161. [Google Scholar] [CrossRef]
- Bachem, M.G.; Zhou, S.; Buck, K.; Schneiderhan, W.; Siech, M. Pancreatic stellate cells—role in pancreas cancer. Langenbeck’s Arch. Surg. 2008, 393, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, I.; Novozhilov, A.S.; Bratus, A.S. Open quasispecies models: Stability, optimization, and distributed extension. J. Math. Anal. Appl. 2020, 481, 123477. [Google Scholar] [CrossRef]
- Bratus, A.S.; Hu, C.K.; Safro, M.V.; Novozhilov, A.S. On Diffusive Stability of Eigen’s Quasispecies Model. J. Dyn. Control Syst. 2014, 22, 1–14. [Google Scholar] [CrossRef][Green Version]
- Volpert, A.I.; Volpert, V.A. Travelling Wave Solutions of Parabolic Systems; Translations of Mathematical Monographs Reprint; American Mathematical Society: Providence, RI, USA, 1994. [Google Scholar]
- Perrings, C.; Mooney, H.; Mark, W. Chapter 1 the Problem of Biological Invasions; Oxford Academic: Oxford, UK, 2009; pp. 1–16. [Google Scholar] [CrossRef]
- Palencia, J.L.D.; González, J.R.; Rahman, S.U.; Redondo, A.N. Regularity, Asymptotic Solutions and Travelling Waves Analysis in a Porous Medium System to Model the Interaction between Invasive and Invaded Species. Mathematics 2022, 10, 1186. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Peng, J.; Su, Z.; Li, Z.; Zhang, B.; Ma, J.; Zhuo, M.; Zou, D.; Liu, X.; et al. Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nat. Cancer 2021, 2, 49–65. [Google Scholar] [CrossRef]
- Mayerle, J. Pancreatic cancer: Why the cell of origin matters. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 279. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8 + cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Huang, Y.H. Life support for transitory exhausted CTLs. Trends Immunol. 2021, 42, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G. Modelling the Dynamics of LCMV Infection in Mice: Conventional and Exhaustive CTL Responses. J. Theor. Biol. 1998, 192, 283–308. [Google Scholar] [CrossRef]
- Baral, S.; Antia, R.; Dixit, N.M. A dynamical motif comprising the interactions between antigens and CD8 T cells may underlie the outcomes of viral infections. Proc. Natl. Acad. Sci. USA 2019, 116, 17393–17398. [Google Scholar] [CrossRef]
- Takano, S.; Fukasawa, M.; Shindo, H.; Takahashi, E.; Hirose, S.; Fukasawa, Y.; Kawakami, S.; Hayakawa, H.; Kuratomi, N.; Kadokura, M.; et al. Clinical significance of genetic alterations in endoscopically obtained pancreatic cancer specimens. Cancer Med. 2021, 10, 1264–1274. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Type | Doubling Time (Days) | Mortality |
|---|---|---|
| K (KRAS) | 200 | |
| P (TP53) | 800 | |
| C (CDKN2A) | 800 | |
| S (SMAD4) | 900 | |
| 140 | ||
| 180 | ||
| 180 | ||
| 750 | ||
| 600 | ||
| 600 | ||
| 120 | ||
| 120 | ||
| 120 | ||
| 500 | ||
| 100 |
| Cell type | K | P | C | S | ||||
| Number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Cell Type | ||||||||
| Number | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| Mutation type, i | K | P | C | S | ||||
| Symbiosis, | 4 | 1 | 1 | 2 | 20 | 24 | 24 | |
| Mutation type, i | ||||||||
| Symbiosis, | 33 | 32 | 32 | 20 | 20 | 20 | 36 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bratus, A.S.; Leslie, N.; Chamo, M.; Grebennikov, D.; Savinkov, R.; Bocharov, G.; Yurchenko, D. Mathematical Model of Pancreatic Cancer Cell Dynamics Considering the Set of Sequential Mutations and Interaction with the Immune System. Mathematics 2022, 10, 3557. https://doi.org/10.3390/math10193557
Bratus AS, Leslie N, Chamo M, Grebennikov D, Savinkov R, Bocharov G, Yurchenko D. Mathematical Model of Pancreatic Cancer Cell Dynamics Considering the Set of Sequential Mutations and Interaction with the Immune System. Mathematics. 2022; 10(19):3557. https://doi.org/10.3390/math10193557
Chicago/Turabian StyleBratus, Alexander S., Nicholas Leslie, Michail Chamo, Dmitry Grebennikov, Rostislav Savinkov, Gennady Bocharov, and Daniil Yurchenko. 2022. "Mathematical Model of Pancreatic Cancer Cell Dynamics Considering the Set of Sequential Mutations and Interaction with the Immune System" Mathematics 10, no. 19: 3557. https://doi.org/10.3390/math10193557
APA StyleBratus, A. S., Leslie, N., Chamo, M., Grebennikov, D., Savinkov, R., Bocharov, G., & Yurchenko, D. (2022). Mathematical Model of Pancreatic Cancer Cell Dynamics Considering the Set of Sequential Mutations and Interaction with the Immune System. Mathematics, 10(19), 3557. https://doi.org/10.3390/math10193557